

Astrocytomas
- A slow-growing astrocytic tumor composed of bipolar “hair-like” (pilocytic) cells.
- Most common glioma in children.
- Associated with tuberous sclerosis, neurofibromatosis type 1 (NF1), and Li-Fraumeni syndrome.
- Optic nerve and chiasm glioma are associated with NF1.
- Often presents with symptoms of increased ICP (headache, nausea/vomiting), vision loss, ataxia, or cranial nerve deficits depending on location.
- Imaging: Cystic mass with a contrast rim-enhancing nidus or mural nodule with minimal vasogenic edema, dorsally exophytic. Most commonly found in the cerebellum.
- Also prefers midline structures such as the brainstem, optic chiasm, hypothalamus, and deep gray matter (basal ganglia).

Pilocytic Astrocytoma
- Also prefers midline structures such as the brainstem, optic chiasm, hypothalamus, and deep gray matter (basal ganglia).
- KIAA1549-BRAF gene fusion is characteristic of this tumor type.

- 90% 10-year overall survival. It can be treated with surgical resection alone, and rarely progresses to malignant glioma.
- Classic patient presentation: Child presenting with increased ICP/ataxia, found to have a cerebellar cystic mass lesion with an enhancing mural nodule.
Subependymal giant cell astrocytoma (SGCA or SEGA)
- WHO grade 1 tumor almost exclusively seen in pediatric patients with tuberous sclerosis (TS) and before the age of 20.
- Seen in 5-15% of patients with TS.
- Often asymptomatic, but when symptomatic presents with obstructive hydrocephalus due to location in the foramen of Monro.
- Imaging: Well-circumscribed, partially-calcified intraventricular contrast-enhancing mass near the foramen of Monro.

Subependymal Giant Cell Astrocytoma
- WHO grade 1 tumor almost exclusively seen in pediatric patients with tuberous sclerosis (TS) and before the age of 20.

- Generally treated initially with mTOR inhibition with everolimus.
- If acute symptomatic or growing, can be treated with surgical resection.
- Tuberous Sclerosis Review: classically presents with seizures, mental retardation, and adenoma sebaceum. Associated with TSC2/tuberin (most cases) or TSC1/hamartin with cortical or subependymal tubers, hamartomas, renal angiomyolipomas, and cardiac rhabdomyomas.
- Generally treated initially with mTOR inhibition with everolimus.
Pleomorphic xanthoastrocytoma (PXA)
- Found in young patients who present with temporal lobe epilepsy.
- Imaging: Supratentorial peripheral cystic and contrast-enhancing mass abutting the leptomeninges with enhancing dural tail sign and scalloping of overlying bone.
- Associated with BRAFV600E mutations and homozygous CDKN2A/B deletions.
- Treated with surgical resection. However, local recurrence and malignant transformation are common so post-operative radiation is indicated for grade 3 tumors.
Adult-type diffuse gliomas
- Astrocytoma, IDH-mutant (grades 2-4)
- Patients present with progressive neurologic symptoms dependent on tumor location and/or with seizures.
- Imaging: T2-FLAIR mismatch sign is often present, with T2 hyperintensity and relative hypointensity on FLAIR sequences.
- MR Spectroscopy will have an elevated choline peak, low NAA peak, and elevated choline:creatinine ratio.
- Pathology:
- Grade 2: mitotic activity absent or low without microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor (homozygous deletion of CDKN2A/B).
- Grade 3: mitotic activity present without microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor.
- Formally known as anaplastic astrocytoma.
- Grade 4: microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor present.
- Glioblastoma, IDH-wildtype (grade 4)
- Formally known as glioblastoma multiforme (GBM).
- The most common and also most destructive of the diffuse gliomas.
- Imaging: Contrast ring-enhancing lesions with significant vasogenic edema. Lesions can also have internal necrosis and can extend through the corpus callosum (butterfly lesion).
- Astrocytoma, IDH-mutant (grades 2-4)

Glioblastoma Multiforme
Left: Axial MRI, T2 FLAIR. Right: T1 w/ contrast. Note the hyperintensity on T1 w/ contrast.

Glioblastoma Multiforme
Left: Axial MRI, T2 sequence. Right: T2 w/ contrast. Note the central necrosis.
")
Glioblastoma Multiforme (GBM)
Left: Axial MRI, T1 w/ contrast. Middle: T2 FLAIR. Right: ADC.
- Genetics:
- IDH-1/2 mutations are associated with secondary GBM arising from a lower-grade glioma.
- Tumors without microvascular proliferation or necrosis can still be classified as “molecular” glioblastoma if genetic testing shows TERT promoter mutation, EGFR gene amplification, or gain of 7/loss of 10 chromosome copy number alterations.
- Pathology: Cells with increased mitotic activity with pseudopalisading necrosis and microvascular endothelial proliferation.
- Genetics:

Glioblastoma Multiforme
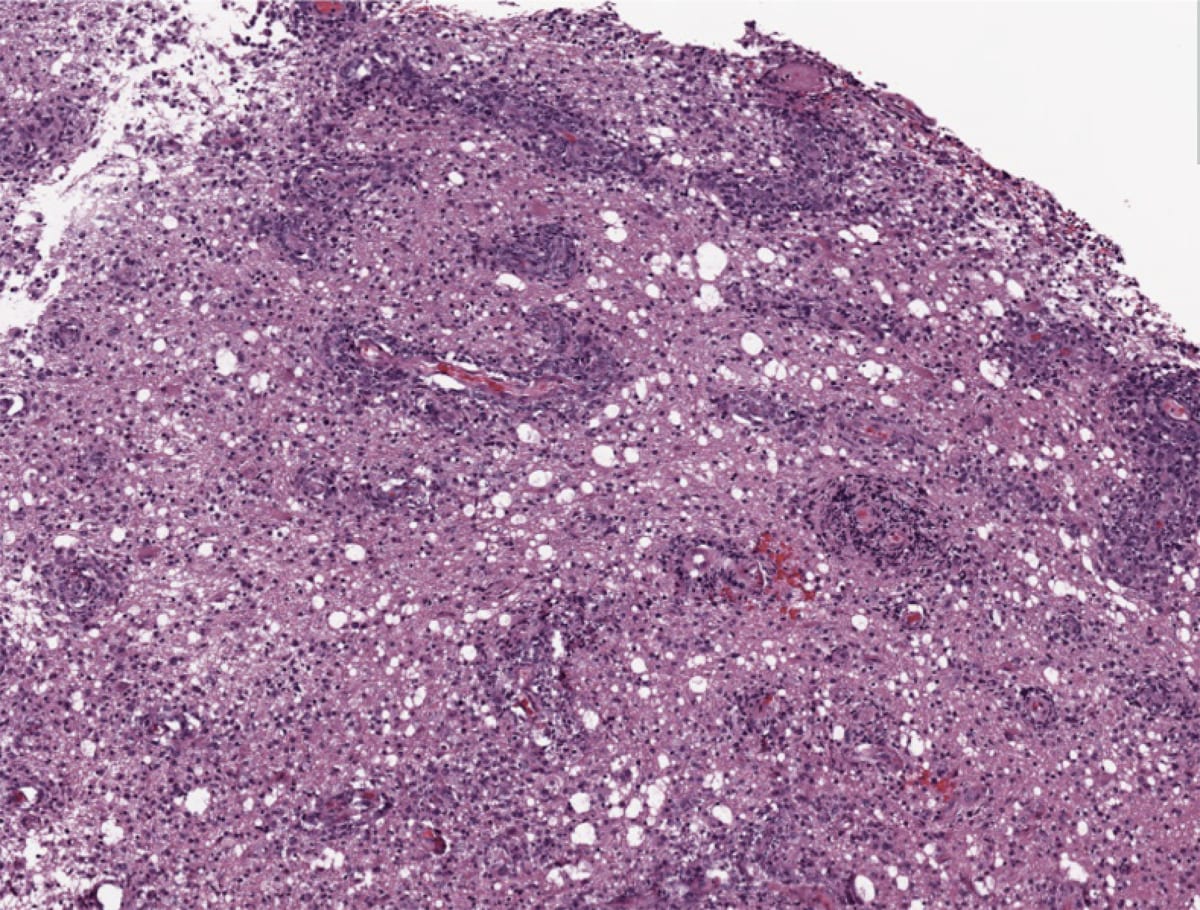
Low-power view showing palisading, or pseudo-palisading, necrosis with a rim of viable tumor-cell nuclei (arrows) lining up around a region of necrotic tumor cells (central pink region).

Glioblastoma Multiforme
Coronal gross cut. Large necrotic mass with increased vascularity and mass effect on the surrounding white matter.
- Treatment: Maximal safe resection followed by intensity-modulated radiation therapy (IMRT) plus concomitant temozolomide (alkylating chemotherapy) followed by adjuvant temozolomide.
- Methylation of the MGMT gene is associated with better treatment response to temozolomide.
- Bevacizumab, a monoclonal antibody that inhibits vascular endothelial growth factor (VEGF) can be used in recurrent or progressive GBM.
- Another alkylating agent, lomustine, is often used as a second-line treatment or in recurrent gliomas.
- Treatment: Maximal safe resection followed by intensity-modulated radiation therapy (IMRT) plus concomitant temozolomide (alkylating chemotherapy) followed by adjuvant temozolomide.

Oligodendrogliomas
- WHO grade 2 or WHO grade 3.
- Associated with 1p/19q co-deletion and IDH mutations;
- 1p/19q co-deletion patients have a better overall prognosis compared to astrocytic tumors which are 1p/19q normal.
- Imaging: Partially calcified T2 heterogeneous, hyperintense subcortical/cortical mass with patchy or minimal contrast enhancement. Most often found cortically in the frontal or temporal lobes.

Attributed by RadsWiki, CC BY-SA 3.0, via Wikimedia Commons, https://upload.wikimedia.org/wikipedia/commons/2/28/Oligodendroglioma_001.jpg, https://upload.wikimedia.org/wikipedia/commons/9/9b/Oligodendroglioma_007.jpg
- Commonly occurs in the 4th or 5th decade of life.
- Pathology: Cells with a “fried egg” appearance with monotonous round nuclei, surrounded by prominent perinuclear halos.
- Treatment: Surgical resection followed by radiation and chemotherapy.

Oligodendroglioma
Neoplastic oligodendrocytes with rounded nuclei, lacking abundant fibrillary processes, and with perinuclear halos giving it a “fried egg” appearance.

Oligodendroglioma
Neoplastic oligodendrocytes with rounded nuclei, lacking abundant fibrillary processes, and with perinuclear halos giving it a “fried egg” appearance.
Ependymal Tumors
- Ependymal tumors are classified based on a combination of factors including anatomic site, histopathology, and molecular features.
- Supratentorial ependymomas
- More frequently in frontal and parietal lobes.
- Imaging: irregular contrast enhancement, often with cysts or calcifications.
- Most common molecular findings: fusion gene involving ZFTA or YAP1.
- Treatment: surgical resection followed by radiation.
- Posterior fossa ependymomas
- Most frequently in the fourth ventricle; can arise in the cerebellopontine angle.
- Clinical presentation related to mass effect/hydrocephalus from fourth ventricular obstruction.
- Headache, vomiting, lethargy; rapid head circumference increase in infants.
- Imaging: homogenous mass within the fourth ventricle.
- Spinal ependymomas
- Spinal ependymoma
- Intradural, intramedullary lesion of the spinal cord.
- Imaging: Contrast-enhancing, well-circumscribed lesion often associated with syrinx.
- Myxopapillary ependymoma
- Intradural, extramedullary lesion of the spinal cord, most often in the lumbar spine.
- Imaging: Contrast-enhancing, well-circumscribed lesion; often described as “sausage-like” in appearance.
- MCYN-amplified ependymoma
- Intradural, extramedullary lesion of the spinal cord, most often in the cervical spine.
- Aggressive compared to other ependymomas.
- Spinal ependymoma
- Subependymomas
- WHO grade 1 tumor most often found incidentally in the ventricles.
- Pathology: Monomorphic cells with round / oval nuclei and salt and pepper chromatin and perivascular pseudorosettes.

Desmoplastic infantile astrocytoma and ganglioglioma
- WHO grade 1, often present before age 2, more often in males.
- Presents with a rapid increase in head size.
- Imaging: large cystic tumor with a peripheral solid component attached to the meninges of frontal or parietal lobes.
- Treatment: surgical resection.
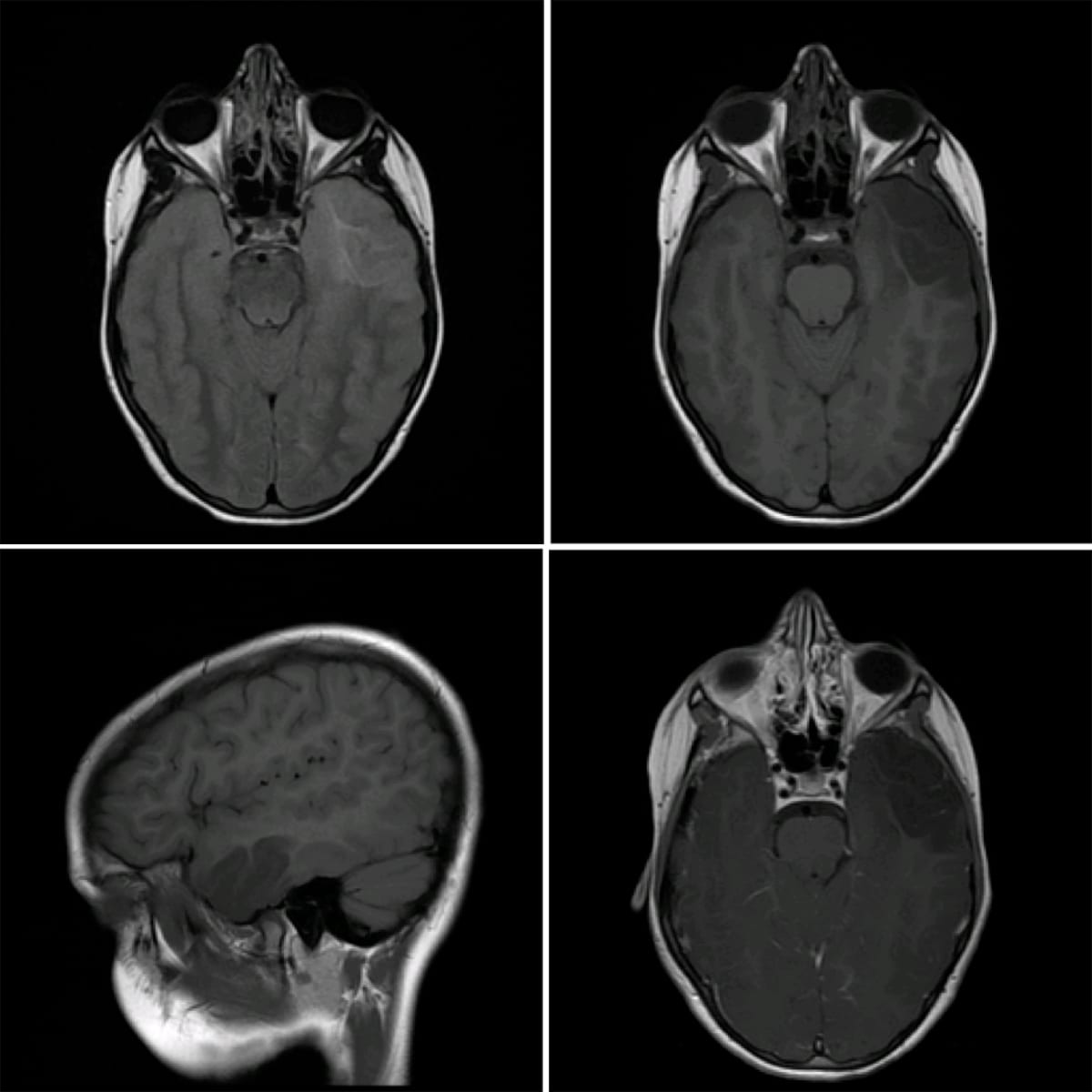
Dysembryonic neuroepithelial tumor (DNET)
- WHO grade 1 tumor arising from cortical gray matter, associated with cortical dysplasia, most often in temporal lobes.
- Characteristically causes temporal lobe epilepsy.
- Imaging: A wedge-shaped cystic cortical mass lesion with a “soap-bubble” appearance, typically non-enhancing.

Dysembryonic neuroepithelial tumor (DNET): Note the “soap bubble” appearance and lack of enhancement.
- Imaging: A wedge-shaped cystic cortical mass lesion with a “soap-bubble” appearance, typically non-enhancing.
Pineal region tumors
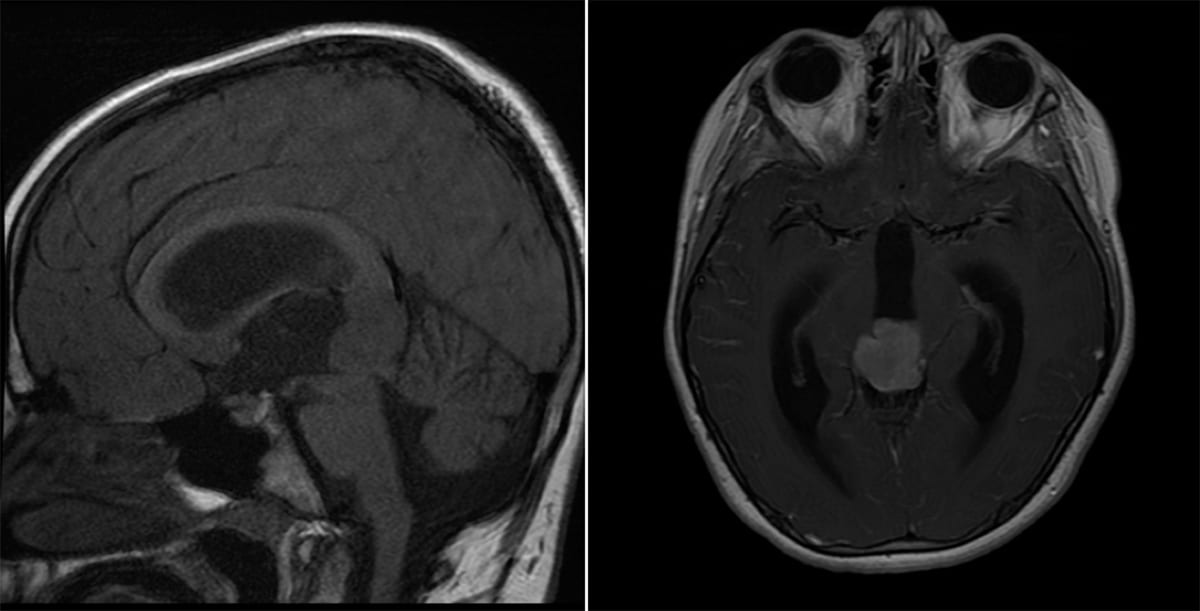
Pineoblastoma
- Imaging: A calcified heterogeneous soft tissue mass in the pineal region with enhancement.

Pineoblastoma - Seen primarily in children.
- These lesions can lead to hydrocephalus secondary to mass effect.
- Pathology: Densely cellular primitive neuroectodermal tumors that often form Homer Wright (medulloblastoma-type) rosettes.
- Treatment: Gross total resection followed by radiation and chemotherapy.
- Imaging: A calcified heterogeneous soft tissue mass in the pineal region with enhancement.
Pineocytomas
- Imaging: Contrast-enhancing midline lesion with irregular borders.
- Patients often present with symptoms of hydrocephalus or mass effect.
- Pathology: Small mature pinealocytes with round/ovoid nuclei forming pineocytomatous rosettes.
- Treatment: Often curative with surgical resection alone.
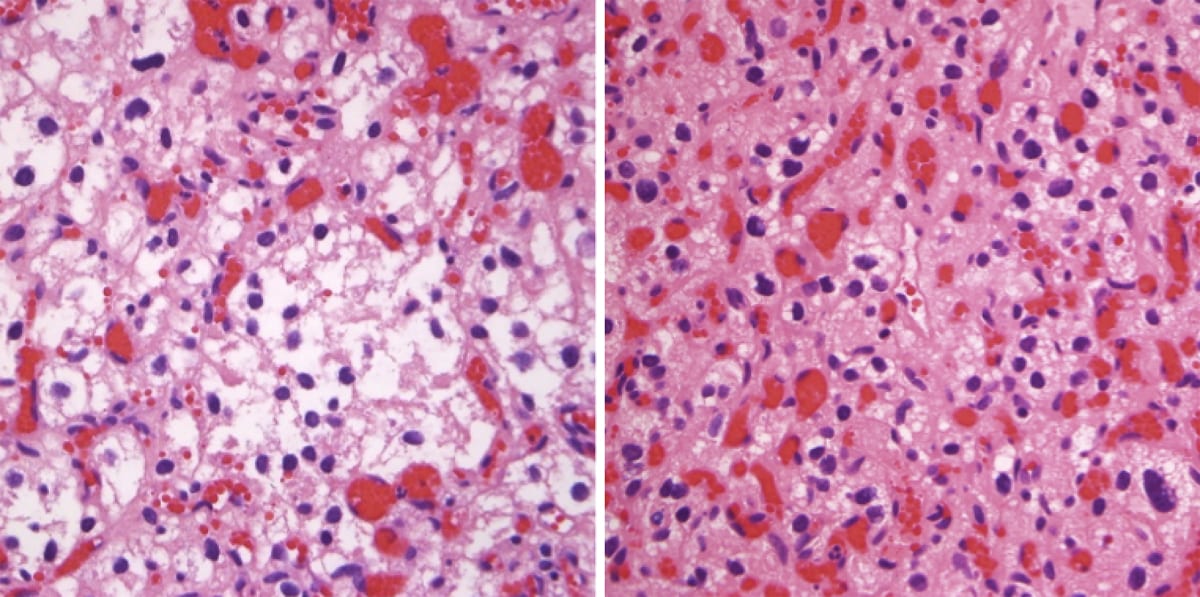
Embryonal tumors
Medulloblastoma
- Medulloblastoma is the most common malignant brain tumor in childhood (20–25% of pediatric CNS neoplasms).
- Associated with Li-Fraumeni syndrome and familial adenomatous polyposis (FAP).
- Imaging: A midline round lesion along the fourth ventricle between the brainstem and cerebellum.

Medulloblastoma
Left: Axial MRI, T2 FLAIR. Right: Coronal MRI, T2 FLAIR.

Medulloblastoma
Left: Axial MRI, T1 w/ contrast. Right: Sagittal MRI, T1 w/ contrast.
- Pathology: Homer-Wright rosettes with no lumen, with necrosis and pseudopalisades.

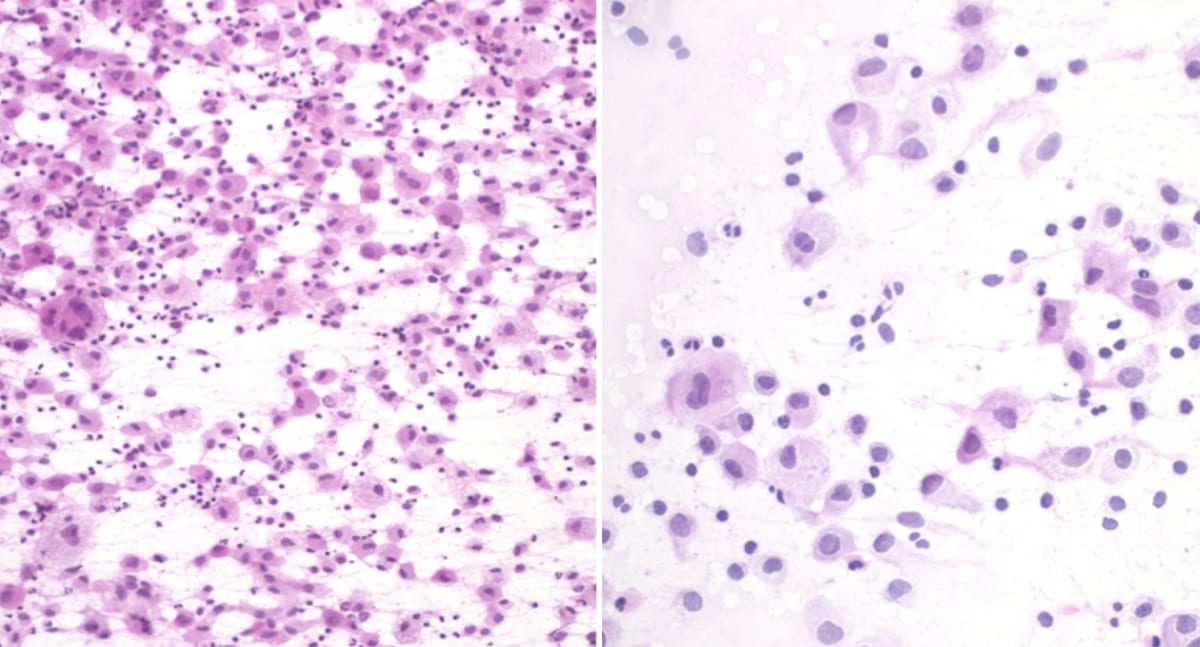
Medulloblastoma
H&E stain with tightly-packed neoplastic cells with round-to-ovoid or “carrot-shaped” nuclei. There is also high mitotic and/or apoptotic activity, and neuroblastic rosettes (Homer Wright rosettes, white arrowheads).

Medulloblastoma
Smear showing hypercellularity with small round blue cells and scant cytoplasm.

Medulloblastoma
Cerebellar mass with mass effect into the 4th ventricle consistent with a medulloblastoma.
- Medulloblastomas can seed and develop disseminated metastatic disease all along the neuraxis.
- Treatment: Gross total resection followed by craniospinal irradiation and chemotherapy.
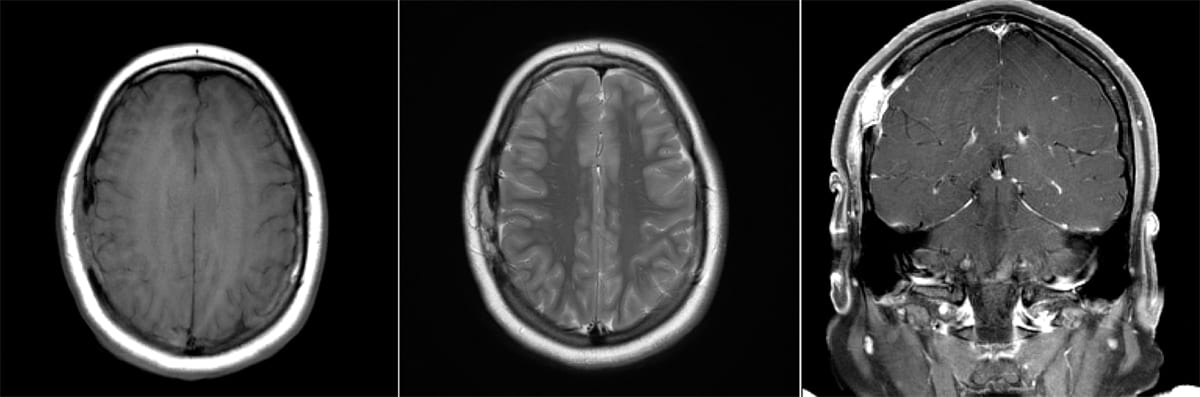
Meningioma
- Imaging: A well-circumscribed dural-based extra-axial diffuse and homogenous enhancing lesion with a “dural tail”.

Meningioma
Left: Axial MRI, T2 FLAIR. Right: T1 w/ contrast (enhancing).

Multiple Meningiomas in NF Type 2
Axial MRI, T1 w/ contrast.

Meningioma
Axial MRI, T1 w/ contrast showing a contrast-enhancing lesion in the left posterior head region.
- Pathology: Calcified concentric psammoma bodies with a “whorled architecture”.

Low power H&E stain of a meningioma showing the whorled architecture of psammoma bodies. - Neurofibromatosis Type 2 (NF2) patients can present with multiple meningiomas.

Multiple Meningiomas in NF Type 2 - Treatment: Observation for asymptomatic low-grade tumors. If symptomatic, resection is performed. Radiotherapy is considered if the meningioma is WHO grade 3.

Schwannoma
- Schwannomas most commonly arise on the CN VII/VIII complex but can arise on any cranial nerve except I and II, which do not have Schwann cells.
- Patients tend to present with sensorineural hearing loss.
- Imaging: Contrast-enhancing mass usually within the internal auditory canal or cerebellar pontine angle.

Acoustic Schwannoma - Pathology: Areas of compact elongated cells with nuclear palisades called Verocay bodies.
- Bilateral acoustic schwannomas are pathognomonic for patients with NF2.

Neurofibroma
- Benign tumors, typically on peripheral nerves.
- Associated with neurofibromatosis type 1 (NF1).
Optic Nerve Glioma
- Typically occurs on CN II and along the visual pathway.
- Astrocytomas, either fibrillary (60%) or pilocytic (40%)
Carcinomatosis Meningitis
- Presents with multiple cranial neuropathies.
Lymphomatous Meningitis
- Presents with multiple cranial neuropathies.
Hemangioblastoma
- Imaging: Contrast-enhancing nodule surrounding a non-enhancing cyst in the posterior fossa with little vasogenic edema.
- Pathology: Large vacuolated stromal cells and variably sized small vascular structures.

H&E stain showing fragments of a neoplasm composed of numerous thin-walled vessels lined with abundant vascular cells, and interspersed vacuolated stromal cells. Occasional stromal cells have large atypical and hyperchromatic nuclei. Additionally, there are focal areas of hemorrhage and hemosiderin deposition. - Commonly seen in patients with Von-Hippel Lindau (VHL) syndrome.
CNS Lymphoma
- Imaging: CNS lymphoma can have a highly variable appearance ranging from diffusely infiltrative, to a solid margin, to lobulated margins. Lesions can have variable enhancement and possibly diffusion restriction. They tend to be located within the basal ganglia or periventricular white matter and can involve the corpus callosum. In immunocompetent patients, lesions are more likely unifocal and are uniformly contrast-enhancing. In immunocompromised patients, lesions may be multifocal and are ring-enhancing.

CNS Lymphoma
Left: T2 FLAIR. Right: T1 w/ contrast.

B Cell Lymphoma
Coronal MRI, T1 sequence w/ contrast.

AIDS Lymphoma
Left: T2 FLAIR. Right: T1 w/ contrast.
- Pathology: Positive staining for B cell marker, CD20.

Low power view CD20 stain showing clusters of lymphocytes. - The elderly and immunocompromised patients are at risk of CNS lymphoma, especially if EBV positive.
- Treatment: Standard initial treatment is combination therapy that includes high-dose methotrexate. In select patients, consolidative therapy with autologous stem cell transplant is performed after initial treatment.
Intravascular lymphoma:
- Presents as a B-cell-driven disease that has multi-organ involvement that may involve the central nervous system.
- If CNS involvement is present, patients present with multiple small brain infarcts.
- Pathology: Lymphoma cells within the blood vessels.
Hemangiomas
- Imaging: Lesion with a “popcorn appearance” with a perimeter of increased signal due to hemosiderin.
- Lesions are at an increased risk of bleeding.
Multiple myeloma
- Lesions will involve bony structures and can lead to compression fractures.
- Imaging: “Salt and pepper” pattern with diffuse heterogeneous lesions of vertebrae.
Langerhans cell histiocytosis
- Due to an uncontrolled monoclonal proliferation of Langerhans cells.
- Pathology: Langerhans cells have large, convoluted, and folded nuclei and are CD1a positive.

H&E stain showing cells with abundant, pale eosinophilic cytoplasm, irregular and elongated nuclei. - More common in children and female predominant.
- Imaging: Lytic lesions in the skull and severe vertebral compression fractures with vertebra plana configuration.

Langerhans Histiocytosis
Germinoma
- Most common pineal region neoplastic lesion.
- Imaging: Solid, contrast-enhancing midline pineal mass near the 3rd ventricle.

Germinoma - Pathology: Small reactive lymphocytes and large neoplastic germ cells with clear cytoplasm. They are histologically similar to cells in the ovaries and testes.


Germinoma
H&E stained, low magnification image showing large round neoplastic cells with clear cytoplasm

Germinoma
Medium power view cKIT stain (tumor marker) showing small reactive lymphocytes and large neoplastic germ cells that stain positively.
- Males are at a much higher risk than females.
- Should be considered in patients who are young and develop diabetes insipidus.
Craniopharyngioma
- Imaging: Partially calcified cystic/solid multi-compartmental suprasellar mass.

Craniopharyngioma
Top left and right: T2 FLAIR. Bottom left and right: T1 w/ contrast. Note hyperintensity with contrast.

Craniopharyngioma
Left: Axial MRI, T2 FLAIR. Right: Sagittal MRI, T1 w/ contrast.
- Derived from Rathke pouch epithelium.

Craniopharyngioma
Encapsulated tumor with well-differentiated non-keratinizing squamous epithelium and papillary fibrovascular stroma.

Craniopharyngioma
Sagittal cut gross specimen with lesion arising from the region of the hypophyseal stalk with both solid and cystic areas.
Pituitary macroadenoma
- Most common suprasellar mass in adults.
- Can lead to compression of the optic chiasm.
- Imaging: A large, enhancing sellar/suprasellar mass.

Pituitary Adenoma
Axial gross pathology. Note the compression of the optic chiasm.

Pituitary Macroadenoma
Coronal (top) and sagittal (bottom) MRI views.

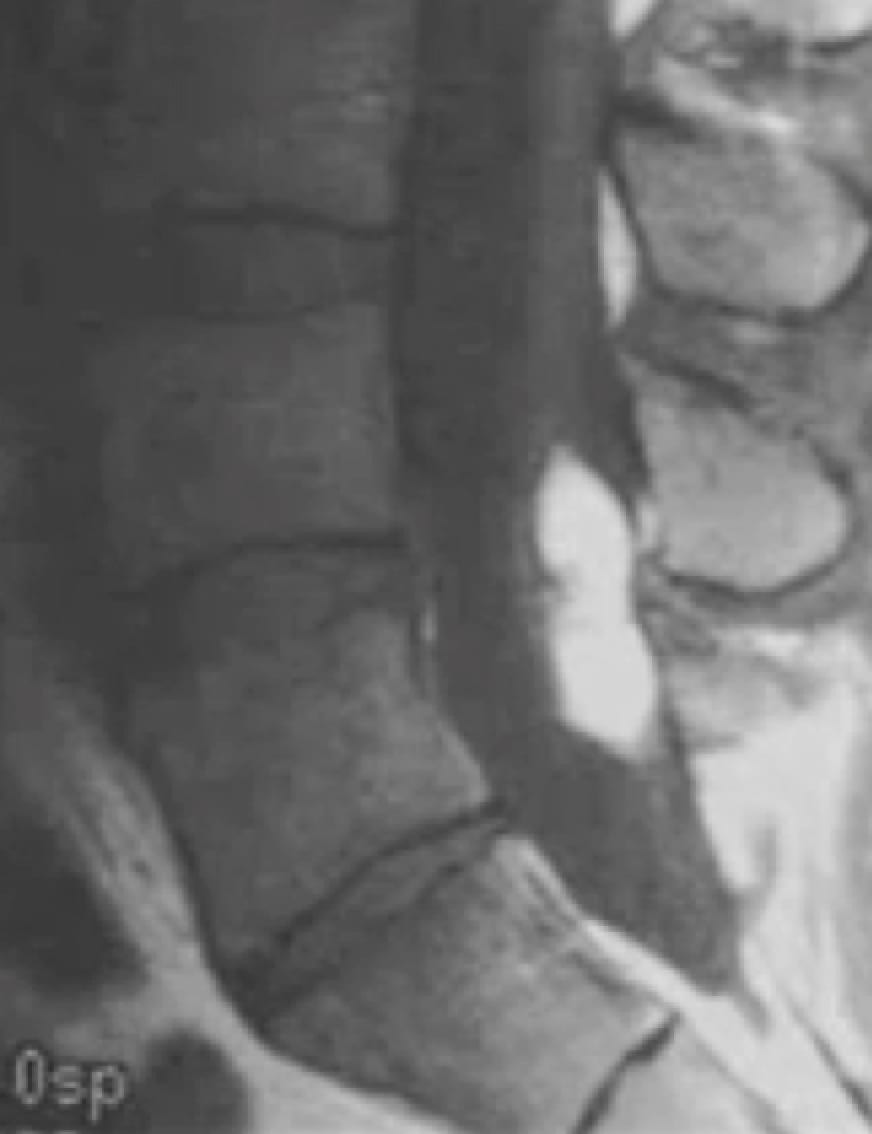
Colloid cyst
- Imaging: A hyperdense non-enhancing epithelial-lined cyst of the foramen of Monro.

Colloid Cyst
Left: Coronal T2 FLAIR MRI. Right: Axial T2 MRI.

Colloid Cyst in the Foramen of Monro
Coronal MRI, T2 FLAIR sequence.
- Lesions are at risk of developing obstructive hydrocephalus.
- Presents with intermittent or persistent headaches due to the increased intracranial pressure from obstruction of CSF outflow.
- The peak age of onset is the third to fourth decade of life.
- Treatment: microsurgically, endoscopically, or with biventricular shunts in nonsurgical candidates.
Epidermoid cyst
- Imaging: An extra-axial mass with a cystic appearance.
- Will be low density and similar to CSF on CT scan, like an arachnoid cyst.
Synovial cyst
- Synovial cysts can form adjacent to any joint throughout the body. If they occur in the spine they can occur in the cervical, thoracic, or lumbar regions.
- Imaging: A well-circumscribed, smooth, extra-dural lesion adjacent to facet joints. Calcification within the cyst can also occur.
- Presenting symptoms include radicular pain and symptoms related to the compression of the adjacent root.
Rathke cleft cyst
- Imaging: Cystic lesion of the pituitary stalk.
- Patients will commonly present with pituitary dysfunction.
Arachnoid cyst
- Arachnoid cysts are extra-axial, congenital space-occupying lesions that displace parenchymal brain tissue and usually are asymptomatic.
- Imaging: A fluid collection that is isodense/isointense to CSF on all sequences (CT, MRI, FLAIR, DWI).

Arachnoid Cyst
Axial MRI, T1 sequence.

Right frontal arachnoid Cyst
Axial MRI, T2 sequence.

Arachnoid Cyst
Axial MRI.
Mucocele
- Imaging: Opacification of the sinus.
- Mucoceles are at risk for abscess formation.
Lipoma
- Imaging: An intradural extramedullary sharply circumscribed homogenous mass .
- If found intracranially they may be near the corpus callosum.
Neurofibromatosis type I (NF-1)
- Also known as von Recklinghausen disease.
- Autosomal dominant disease related to the NF1 gene, which codes for neurofibromin.
- Non-neurologic findings include café-au-lait spots, Lisch nodules, skeletal abnormalities, and axillary freckling.
- Neurologic lesions:
- Optic nerve gliomas
- Patients should have yearly eye exams.
- Typically found in the first 6 years of life.
- Small asymptomatic gliomas can be observed, as they may regress. Larger, symptomatic lesions are treated with chemotherapy.
- Neurofibromas
- Neurofibromas occur on cutaneous branches of peripheral nerves, including neuronal roots and plexuses. They tend to be slow-growing and can remodel the surrounding bone.
- Lesions encase the nerve and are contrast-enhancing.
- Optic nerve gliomas

Lisch nodules
Brown-yellow melanocyte aggregates within the iris consistent with Lisch nodules seen with Neurofibromatosis type 1
Attributed by Dimitrios Malamos, CC BY 4.0, via Wikimedia Commons
https://commons.wikimedia.org/wiki/File:Lisch_nodules.JPG

Cutaneous Neurofibromas
Patient with Neurofibromatosis type 1 with innumerable cutaneous neurofibromas
Attribution by Klaus D. Peter, Wiehl, Germany, CC BY 3.0 DE, via Wikimedia Commons
https://commons.wikimedia.org/wiki/File:Neurofibroma02.jpg

Cafe Au Lait Spot
By Original uploader was Accrochoc at fr.wikipedia – Transferred from fr.wikipedia; transferred to Commons by User:Andreas Werle using CommonsHelper., CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=4513102
Tuberous Sclerosis
- Autosomal neurocutaneous disorder related to two tumor-suppressor genes: TSC1 (9q34) which encodes hamartin and TSC2 (16q13) which encodes tuberin.
- Imaging will show cortical tubers which can be foci for seizures.
- Seizures commonly occur secondary to cortical tubers.
- Non-neurologic findings include ash-leaf spots, facial angiofibromas, angiomyolipomas, and retinal hamartomas.
- Intellectual disability can occur in severe cases.
- 5-10% of patients can develop subependymal giant cell astrocytomas.

Mnemonic: Think TUBERRRS: Tsc2, U(ventricular), Bulk removal, Eosinophilic Rhabdomyosarcoma/Renal angiomyolipoma/Retinal Hamartomas, Seizures
Leptomeningeal carcinomatosis
- Imaging: Contrast enhancement along the leptomeninges.
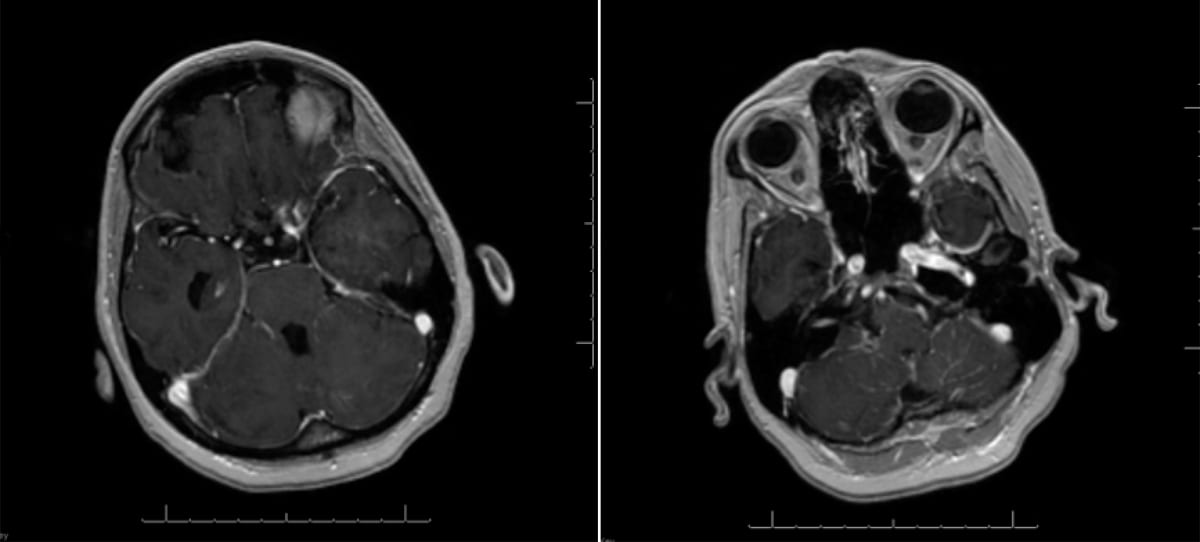
- When on the differential, patients should have the whole neuraxis imaged to look for additional lesions as well as CSF analysis.
- Obstruction of CSF re-absorption from the malignancy can lead to communicating hydrocephalus. Patients will present with severe headaches, nausea, and vomiting.
- Symptoms can improve with large volume lumbar puncture or EVD placement.
- Symptoms often precede cancer diagnosis and thus patients require cancer surveillance for a minimum of 2 years after symptom onset.
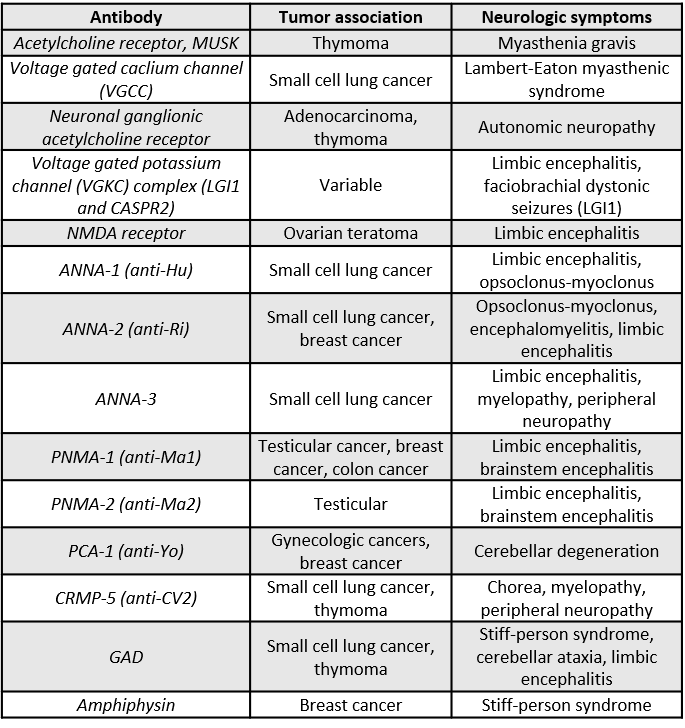
Limbic encephalitis
- Most associated with SCLC. testicular germ cell tumors or ovarian teratoma.
- Several antibodies are related to paraneoplastic disorders including anti-Hu, voltage-gated potassium channels, NMDA receptors, etc (see table below).
- Slightly more than half of patients with autoimmune limbic encephalitis have detectable CNS antibodies.
- Patients will present with psychiatric/cognitive abnormalities, seizures, facial and limb dyskinesias, and autonomic instability.
- Imaging will show abnormalities in cortical and subcortical regions.
- Young women who present with this syndrome should be evaluated for ovarian teratoma.
Paraneoplastic cerebellar degeneration
- Presents with a subacute progression of ataxia, nystagmus, and dysarthria.
- Gynecological (ovarian, uterine, breast) and small-cell lung cancers are common causes.
- Pathology: Profound loss of Purkinje cells.
- MRI: often normal, may have some enhancement near the cerebellum, and may have cerebellar atrophy.
- Recovery is slow or partial (postmortem studies show permanent loss of Purkinje cells).
Lambert-Eaton Myasthenic Syndrome (LEMS)
- Presents with proximal weakness and fatigability, impotence, and ptosis.
- May be associated with paraneoplastic encephalomyelitis and cerebellar degeneration.
- 70% of patients have cancer (most often SCLC).
- For more information see the Neuromuscular Junction Disease chapter.
Other paraneoplastic syndromes
- Myasthenia gravis (thymoma)
- Subacute sensory neuronopathy
- Opsoclonus-myoclonus syndrome
- Presents with rapid, high-amplitude conjugate eye movements (opsoclonus), myoclonus, ataxia, and ataxia.
- Related to neuroblastoma in children, and breast, ovarian, or small cell lung cancer in adults.

Neuroblastoma with Rosettes
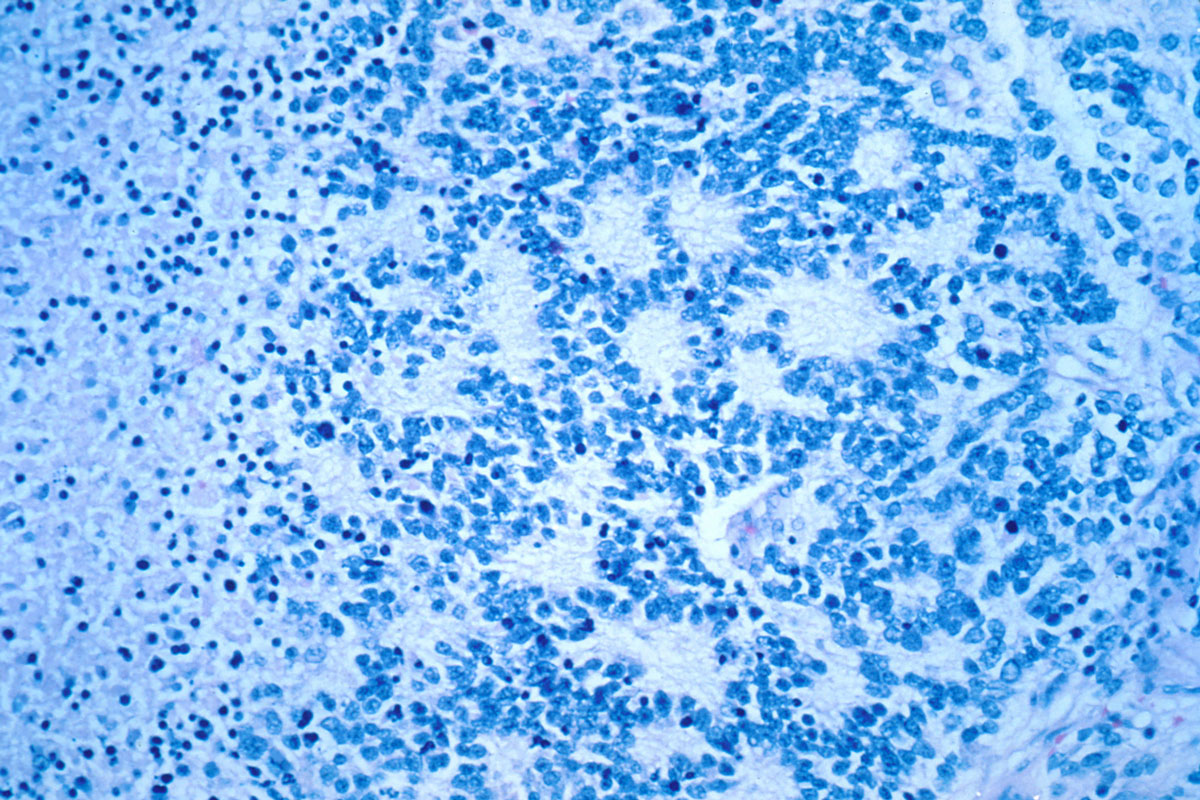
- Stiff-person syndrome
- Association with anti-amphiphysin (paraneoplastic) or anti-GAD (more often autoimmune than paraneoplastic).
- Neuro-myotonia
- Dysautonomia
- Myelopathy
Log in to View the Remaining 60-90% of Page Content!
Important: If you signed up after 1/1/2026, or if you opted to migrate your old account to the new & improved platform (same great content, better experience), please log in at nowyouknowmed.com
