


- Typical findings in CSF include an elevated protein, elevated CSF IgG index, oligoclonal bands in the CSF which do not present in the serum, and possibly slight elevation of WBC count (lymphocytic predominant).
- MRI imaging with contrast is the most important imaging modality in the workup of possible MS.
- Demyelinating lesions related to MS are ovoid, >3 mm in diameter, and are typically located in particular regions: periventricular (Dawson’s fingers), juxtacortical, cortical, or spinal cord.

Multiple Sclerosis Plaque
Pericallosal MS plaque on T2 FLAIR (arrow).

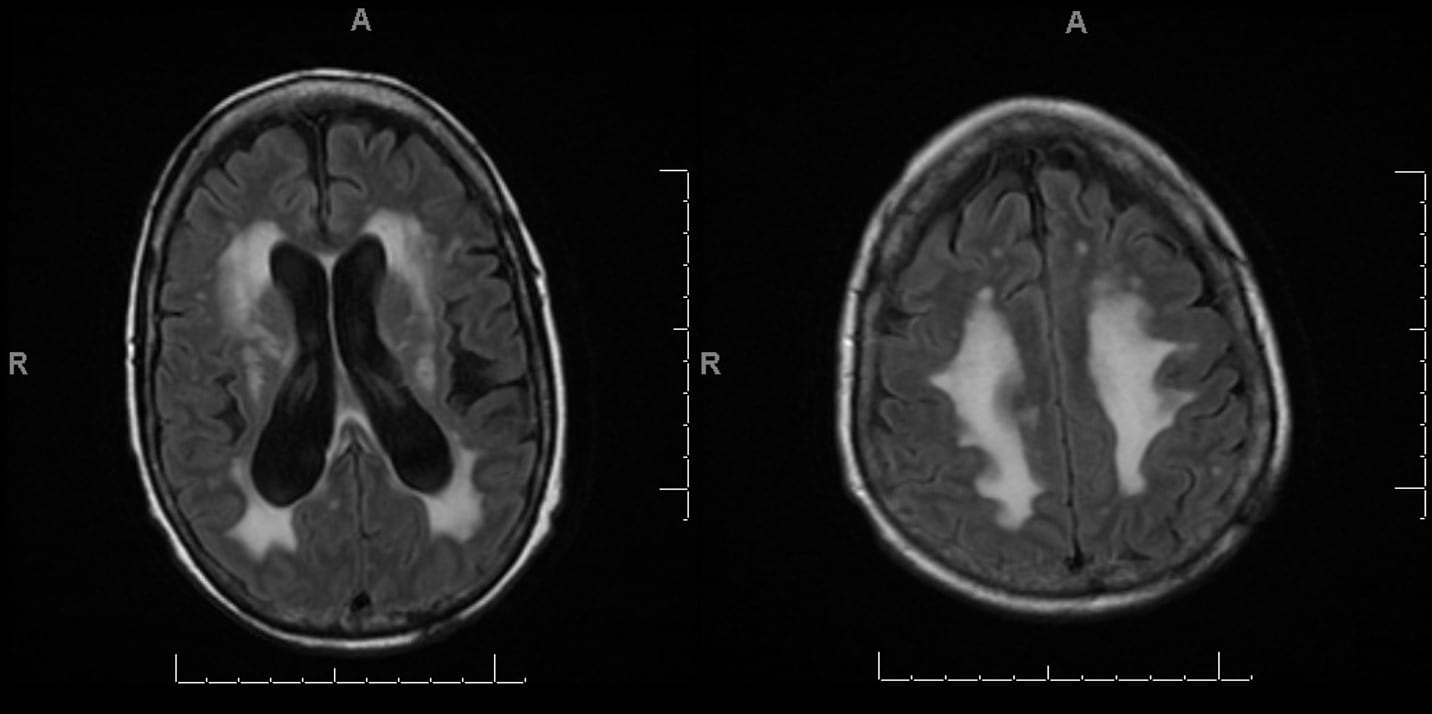
Dawson’s Fingers in Multiple Sclerosis
Sagittal T2 FLAIR MRI.

Dawson’s Fingers in Multiple Sclerosis
MRI T2 flair sequence, sagittal cut.

Multiple Sclerosis Plaques
Axial MRI, T2 FLAIR.
Optic Neuritis (ON)
- Presents with monocular decreased visual acuity, loss of color vision, and an afferent pupillary defect (APD) due to a demyelinating lesion of the optic nerve.
- Pain is also usually present with extraocular movements.
- Children with ON are more likely to present with headache and bilateral symptoms.
- Can be the initial presentation of multiple sclerosis or neuromyelitis optica.
- A funduscopic exam will show a swollen optic disc.
- Optical coherence tomography (OCT) will show thinning of the retinal nerve fiber layer weeks to months after an acute attack.
- Visual evoked potentials (VEPs) of the affected eye will show a prolonged P100 latency, even in patients with distant histories of ON with no residual visual deficits.
Transverse myelitis (TM)
- Presents with myelopathic findings (weakness, hyperreflexia, sensory symptoms, and occasionally bladder/bowel dysfunction).
- Lhermitte’s sign, an electric shock sensation down the neck/spine with neck flexion or extension) may also be present.
- MRI will show a T2/FLAIR hyperintense lesion involving the spine +/- contrast enhancement based on the lesion’s acuity.
- TM can be the initial presentation of demyelinating disease. However, it is also important to exclude infectious, rheumatologic, postvaccination/postinfectious, and nutritional mimics of demyelinating disease.
- If felt to be truly inflammatory or idiopathic TM once other etiologies are ruled out, then initiation of steroids is indicated.

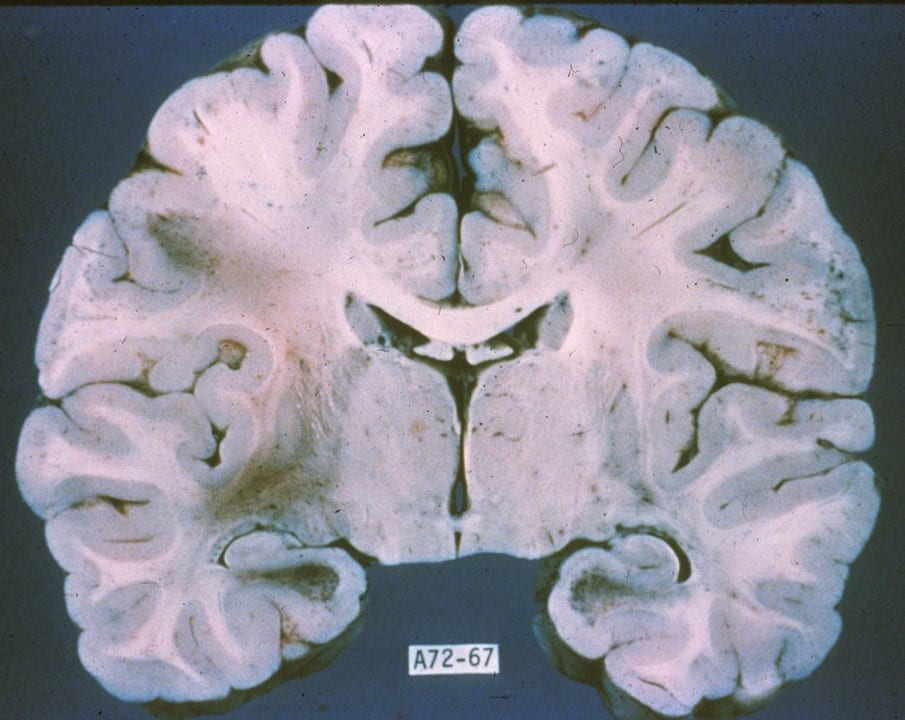
- On gross anatomy, there will be grey plaques within white matter representing the loss of myelinated tissue.

Multiple Sclerosis Plaque
Note the well-demarcated periventricular tan-gray patches of demyelination (arrow).

Multiple Sclerosis Plaque
Plaques are often present adjacent to the lateral angles of the lateral ventricles (pictured), optic nerves, the floor of the fourth ventricle, brain stem, and spinal cord.

Multiple Sclerosis Plaque
Well-demarcated tan-gray patches of demyelination.
- Microscopic analysis can gauge the chronicity of a demyelinating plaque.
- Active plaques will have a destruction of oligodendroglia while sparing neurons and their axons, inflammatory infiltrates of the parenchyma and in the perivascular space, as well as macrophages containing myelin debris.

Active MS Plaque
H&E stain, low power view, showing an active demyelinating plaque with perivascular lymphocytes, macrophages, and reactive astrocytes (arrows).

Multiple Sclerosis Plaque
H&E stain, Low power view, showing sharply demarcated hypocellular plaques.

Multiple Sclerosis
Medium power, H&E stain, brain biopsy showing inflammatory infiltration of the perivascular space
- During the recovery phase, recruited oligodendrocyte precursor cells remyelinate active plaques leading to reduced myelin density and thin myelin sheaths.
- Almost half of chronic MS plaques will show some degree of remyelination.
- Remyelinated plaques are also known as “shadow plaques”.
- Chronic inactive plaques have sharply demarcated hypocellular plaques with myelin loss and glial scar formation via astrocytes.
- During the recovery phase, recruited oligodendrocyte precursor cells remyelinate active plaques leading to reduced myelin density and thin myelin sheaths.

Chronic Multiple Sclerosis Plaque
Luxol fast blue stain, low-power view, showing decreased staining of a chronic demyelinated plaque (arrow).

Hypocellular Multiple Sclerosis Plaque
H&E stain, Low power view, showing a sharply-demarcated hypocellular plaque.

Hypocellular Multiple Sclerosis Plaque
H&E stain, Low power view, showing sharply-demarcated hypocellular plaques.
MS variants
Tumefactive MS (TMS):
- Presents with a large (>2 cm) demyelinating lesion with incomplete ring enhancement, mass effect, minimal surrounding edema, and low cerebral blood volume.
- Imaging may be difficult to differentiate from a brain tumor, ADEM, or abscess.
- Tumors will often have more significant surrounding edema and higher cerebral blood volume than TMS.
- A biopsy is sometimes required to confirm the diagnosis.
- Imaging may be difficult to differentiate from a brain tumor, ADEM, or abscess.
- Pathology: Foamy macrophages containing phagocytosed myelin debris.
- Presents with a large (>2 cm) demyelinating lesion with incomplete ring enhancement, mass effect, minimal surrounding edema, and low cerebral blood volume.
Marburg variant disease (Acute/fulminant Multiple Sclerosis)
- A fulminant form of MS with large tumefactive lesions, associated with a high mortality rate.
- It is relatively unresponsive to therapy but high-dose steroids and PLEX may help.
- Pathology will show more macrophage infiltrates and necrosis than typical MS.
- A fulminant form of MS with large tumefactive lesions, associated with a high mortality rate.
Balo’s concentric MS
- Imaging will show concentric alternating bands of preserved myelination with alternating areas of demyelination. This is also described as “onion bulbs” when seen pathologically.

Schilder’s disease:
- Seen primarily in childhood.
- Lesions will be large in size, contrast-enhancing, and usually involve the centrum semiovale.
- Formally known as Devic disease, NMOSD is an inflammatory CNS disorder distinct from MS, most commonly associated with antibodies to aquaporin-4 (APQ4).
- Core clinical characteristics:
- Optic neuritis (longitudinal/chiasmal lesion)
- Longitudinally extensive transverse myelitis (LETM) is defined by a single lesion involving 3 or more vertebral segments.
- Brainstem/diencephalic lesions.
- Area postrema syndrome: Hiccups, nausea, and vomiting
- Antibodies for aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG) should be checked in any patient when NMO is suspected.
- AQP4 is a water channel localized in the CNS at the endfeet of astrocytes.
- MOG is found on oligodendrocytes.
- Unlike MS, oligoclonal bands are often absent from the CSF.
- Pathologic findings include perivenous infiltration of lymphocytes, inflammation, and demyelination.
- Treatment:
- Acute therapy: High dose steroids, then PLEX or IVIG if symptoms are steroid-resistant.
- Long-term management:
- IVIG
- Immunosuppressants (azathioprine, mycophenolate)
- Monoclonal antibodies (if anti-AQP4 antibody positive)
- The disease-modifying agents for multiple sclerosis have not demonstrated effectiveness for NMOSD.

Asymmetric Thoracic Cord Demyelination
In a patient with neuromyelitis optica (NMO)

Lumbar Conus Medullaris Plaque in Neuromyelitis Optica
Sagittal MRI spine. Note the longitudinally extensive hyperintense lesion in the distal segment of the spinal cord.
- Also known as postinfectious encephalomyelitis.
- A monophasic autoimmune illness that presents abruptly with headaches, fever, and widespread CNS demyelination and occurs primarily in children and young adults.
- Symptoms usually resolve within a week without residual neurologic deficits. Children are more likely to make a complete recovery than adults.
- Disease onset is 7 to 14 days after environmental exposure.
- Most commonly occurs after viral upper respiratory infections but can occur after immunizations as well.
- MRI brain with contrast will show multiple bilateral hyperintense T2-weighted lesions with poor margins and variable enhancement.
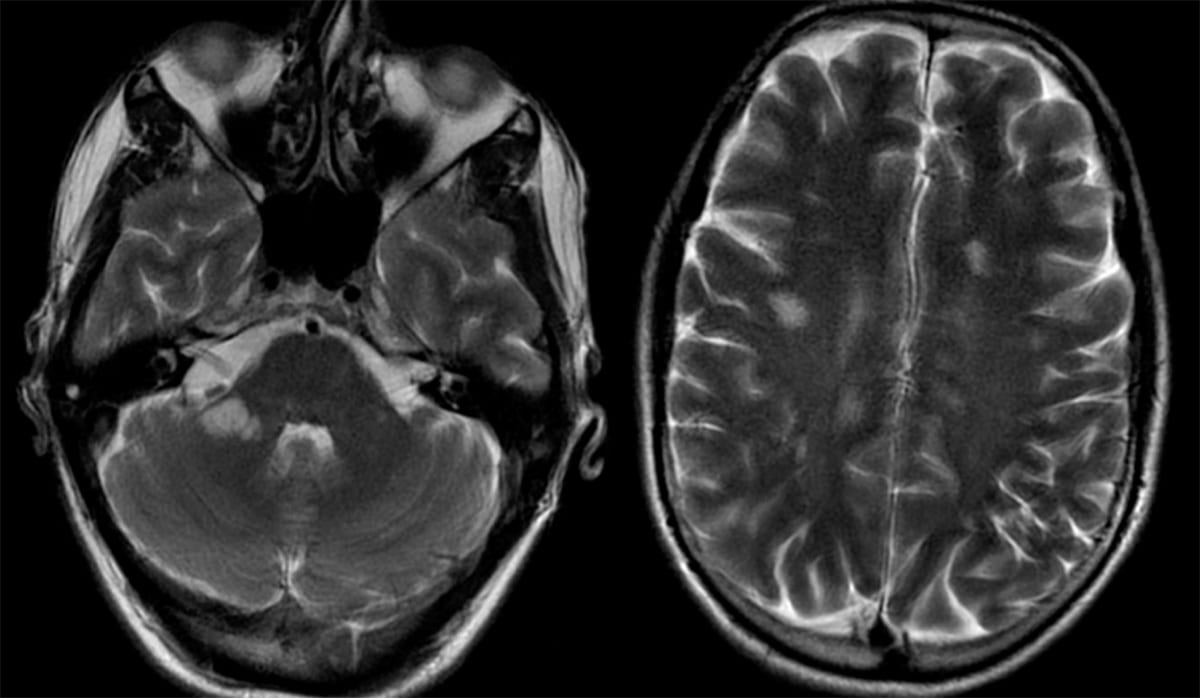
- Pathologic findings include perivenous infiltration of lymphocytes, inflammation, and demyelination.

Caudal Medulla in ADEM
Axial section of the lower medulla shows several overlapping lucent lesions, giving it a moth-eaten appearance.

Acute Disseminated Encephalomyelitis
Luxol fast blue stain, low power view, showing several pale demyelinated areas in the cortical white matter.
- Usually fatal infection of oligodendrocytes caused by reactivation of latent JC polyomavirus.
- Patients will present with subacute progressions of encephalopathy, focal motor deficits, ataxia, diplopia, and/or hemianopia.
- The CNS infection is established via hematogenous dissemination of the latent virus in the kidneys which then crosses the blood-brain barrier.
- Occurs in patients with impaired immune systems: HIV, leukemia, and those on immunosuppressive agents (chemotherapy/monoclonal antibodies).
- For JC-positive patients, natalizumab has a high risk of causing PML when treatment is greater than 2 years.
- Patients on immunosuppressive medications at the time of diagnosis should have their medications discontinued and then consider plasma exchange to remove the drug from the circulation.
- Patients with HIV-related PML should be started on HAART therapy.
- Restoration of immune function may result in immune reconstitution inflammatory syndrome (IRIS), which presents with worsening neurologic symptoms and contrast enhancing cerebral edema .
- IRIS occurs 3-6 weeks after treatment for PML and should be treated with high-dose IV steroids followed by an oral slow taper regimen.
- Restoration of immune function may result in immune reconstitution inflammatory syndrome (IRIS), which presents with worsening neurologic symptoms and contrast enhancing cerebral edema .
- Imaging will show non-enhancing FLAIR lesions preferentially involving the white matter without mass effect.
")
Progressive Multifocal Leukoencephalopathy (PML)
Left: Axial T1. Middle: T2 FLAIR. Right: T2 with contrast with extensive left parietal white matter hyperintensity. Enhancement, which is seen in this case, is atypical and more commonly seen in cases of PML induced by AIDS or natalizumab.
")
Progressive Multifocal Leukoencephalopathy (PML)
Axial T2 FLAIR showing hyperintensity of the left parietal region with involvement of the subcortical U-fibers. There is some mass effect present.
")
Progressive Multifocal Leukoencephalopathy (PML)
Axial FLAIR MRI with multiple areas of hyperintensity involving the medial temporal lobes, the right occipital lobe, and the splenium of the corpus callosum.
- Pathology:
- Demyelination . Immunohistochemistry staining for the JC virus is considered the gold standard for diagnosis.
- JC virus PCR titers can also be used for diagnostic purposes and should be performed before biopsy if clinical suspicion is high.
- Also known as osmotic demyelination syndrome, this disease is due to the rapid correction of hyponatremia.
- This rapid correction leads to the demyelination of the base of the pons.
- Extrapontine demyelinating lesions are infrequent but when present they are seen at cortical gray-white matter junctions.
- Seen in alcoholics, end-stage renal disease, chronically malnourished patients, and patients undergoing a hepatic transplant.
- Patients present acutely with AMS, hypotension, and hypoventilation with progression to pseudobulbar palsy and quadriparesis. Severe cases can progress to a “locked-in” syndrome.
- While there are no available therapies, many patients improve over time.

- Inherited leukodystrophies are a heterogeneous group of inherited, metabolic demyelinating neurodegenerative diseases of the central nervous system.
- Patients with leukodystrophy commonly have an initial period of normal development followed by progressive neurological deterioration. Common symptoms include bilateral and symmetric spasticity, weakness, and ataxia.
- Cases can present at any age, ranging from infancy to adulthood.
- Some leukodystrophies also present with demyelination of peripheral nerves; such as Metachromatic leukodystrophy and Krabbe disease.
- MRI imaging can be variable but it will universally show changes in the cortical white matter, appreciated as T2 sequence hyperintensity. Particular dystrophies may also have white matter contrast enhancement. If only particular regions are involved, this can point you in the right direction to a particular disease.
- Treatment: Adrenoleukodystrophy, metachromatic leukodystrophy, and Krabbe’s disease can be treated with stem cell transplantation.
Metachromatic leukodystrophy
- An autosomal recessive disease due to a deficiency of arylsulfatase A enzyme from a mutation in the ARSA gene.
- Can present in infancy, childhood/juvenile, or adulthood depending on the severity of the gene dysfunction.
- Infants present with vision loss, spastic ataxia, and seizures. Death usually occurs in 2-4 years after disease onset.
- Juvenile onset has similar symptoms but a slower rate of progression.
- Adults present with behavioral change, psychosis, and dementia.
- Imaging will show extensive white matter demyelination
- A demyelinating sensorimotor polyneuropathy is also present in almost all cases.
X-linked Adrenoleukodystrophy
- Due to an X-linked mutation in the gene ABCD1, which is responsible for the function of peroxisomes. The mutation leads to impaired very-long-chain fatty acid (VLCFA) oxidation.
- Blood and urine studies can show increased levels of VLCFAs.
- Within the same family, the same genetic mutation can have variable phenotypes.
- Presents with adrenal failure, testicular atrophy, hyperactivity, ataxia, vision/hearing loss, and seizures.
- Adrenal insufficiency is seen in 50-80% of patients and can cause hyperpigmentation.
- Intermittent adrenal testing may be needed as well as corticosteroid supplementation during times of stress/illness.
- Imaging will show posterior predominant white matter demyelination with sparing the frontal lobes and contrast enhancement.
- Adrenomyeloneuropathy (AMN) variant:
- Primarily affects the spinal cord leading to lower extremity weakness and spasticity.
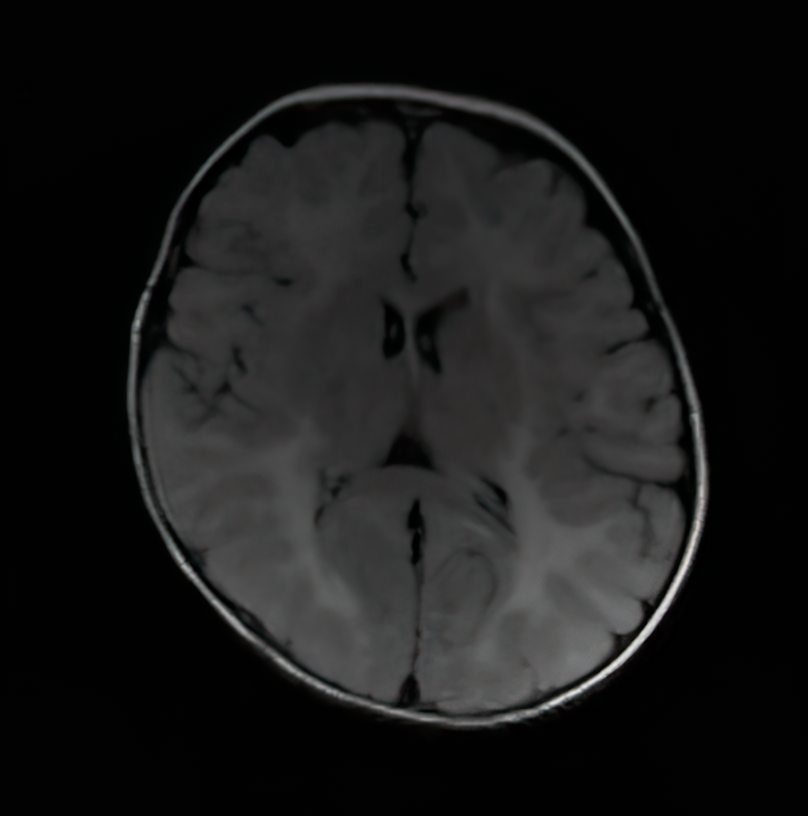
Krabbe disease
- Due to a deficiency of the enzyme galactocerebrosidase.
- The infantile form will present with an exaggerated startle, fevers, behavioral regression, optic atrophy, and hypertonicity. Death usually occurs within a few years of diagnosis. There are juvenile and adult forms as well which are less progressive than the infantile form.
- Seizures are frequent.
- Imaging will show white matter changes .
- Pathology will show large globoid cells which are PAS-positive multinucleated macrophages with galactocerebroside present.
Log in to View the Remaining 60-90% of Page Content!
Important: If you signed up after 1/1/2026, or if you opted to migrate your old account to the new & improved platform (same great content, better experience), please log in at nowyouknowmed.com

{kind=link}