

Peripheral neuropathy is a broad term referring to any disorder of the peripheral nerves and therefore has many causes and many interesting pathologies. As such, this is something the average neurologist sees countless times, and consequently is a high-yield topic on many examinations such as the shelf, RITE®, and board exams!
Authors: Brian Hanrahan MD, Steven Gangloff MD
What you need to know:
Peripheral neuropathy is a general term that can be used to describe polyneuropathy, radiculopathies, and mononeuropathies.
Polyneuropathies
- Polyneuropathy is defined as a neuropathic disease that involves many nerves in a generalized distribution.
- Causes of peripheral polyneuropathy can be either axonal and/or demyelinating.
Small fiber polyneuropathy
- Presents with burning/tingling pain, usually felt to be more significant while trying to go to sleep.
- Causes of small fiber neuropathy are diverse but include diabetes, B12 deficiency, chronic alcohol use, autoimmune diseases (i.e. Sjögren syndrome), amyloidosis, chemotherapeutic agents, HIV, leprosy, and Fabry disease.
- Diagnostic testing:
- Skin biopsy to evaluate small fiber density.
- Quantitative sudomotor axon test (QSART) to evaluate postganglionic sympathetic sudomotor axons.
- It is abnormal in 75% of patients with distal small fiber neuropathy.
- Patients with small fiber neuropathy can also have concurrent autonomic neuropathy.
- Further details of small fiber neuropathy are outlined in the Pain Syndromes chapter.
Note:
EMG/Nerve conduction studies performed on patients with solely small fiber neuropathies will be normal.
Acquired Neuropathies
Systemic disease causes of neuropathy
Diabetes
- Responsible for one-third of all neuropathies (most common cause).
- Can present as a small fiber polyneuropathy, large fiber peripheral polyneuropathy, multifocal neuropathy, mononeuropathy, and/or lumbosacral plexopathy.
- Commonly manifests as a distal symmetric polyneuropathy, typically in a “stocking-glove” distribution.
- EMG testing will show axonal pathology.
- Pathology: Thickened blood vessels with basement membrane reduplication.
- Patients should be screened with a hemoglobin A1c level, fasting glucose, or a 2-hour glucose tolerance test.
Critical illness neuropathy
- Presents with distal axonal sensorimotor polyneuropathy which leads to limb weakness or difficulty weaning off of artificial ventilation.
- Sepsis, SIRS, and multiorgan failure are risk factors.
- Patients with critical illness neuropathy may also have concurrent critical illness myopathy.
- Diagnostic studies may include EMG showing spontaneous activity (i.e. fibrillations) and nerve conduction studies (NCS) showing normal velocity and amplitude reduction.
- Symptoms can take months to years to recover and one-third of patients never walk independently again.
B12 deficiency
- Seen in patients with poor diets: Chronic alcoholism, post-gastric bypass, and nitrous oxide abuse.
- Strict vegan diets may also lead to this deficiency, but this would take years to develop.
- Other causes include atrophic gastritis, Crohn’s disease, celiac disease, and enteritis.
- Symptoms are generally symmetrical and may present as peripheral neuropathy, subacute combined degeneration, or even neuropsychiatric conditions.
- If B12 levels are borderline abnormal, you should check methylmalonic acid and homocysteine levels if the concern is high.
- See the Toxicology chapter for more details.
Copper deficiency
- Presents similarly to B12 deficiency.
- Seen in patients with a malabsorption disorder or from excessive zinc intake.
- May also be seen in genetic diseases such as Menke’s disease.
Connective tissue disease
- Peripheral neuropathy can be seen in patients with polyarteritis nodosa, rheumatoid arthritis, Sjögren’s syndrome, systemic sclerosis, giant cell arteritis, lupus, and Wegener’s granulomatosis.
HIV
- HIV-related distal symmetric polyneuropathy is the most common neurological complication of HIV, affecting 50% of patients.
- Concurrent treatment with nucleoside reverse-transcriptase inhibitors (NRTIs) like didanosine, zalcitabine, and stavudine increases the risk of HIV-related neuropathy.
Hypothyroidism
- Chronic hypothyroidism can present with mono- or polyneuropathy due to compression of nerves.
MGUS/Multiple myeloma
- Presents as a chronic demyelinating disorder that can be similar phenotypically to those with CIDP.
Cryoglobulinemia
- Seen in about 28-50% of hepatitis C patients.
POEMS Syndrome
- POEMS is an acronym for Polyneuropathy, Organomegaly, Endocrinopathy, M protein, and Skin changes.
- Occurs due to high levels of serum vascular endothelial growth factor (VEGF) and low levels of erythropoietin.
- Patients develop a progressive symmetric, length-dependent sensorimotor polyneuropathy.
- Organomegaly presents as hepatomegaly and/or splenomegaly.
- Possible endocrinopathies are diabetes mellitus, hypogonadism, and hypothyroidism.
- “M protein” represents the production of monoclonal proteins. This can be secondary to MGUS, plasmacytoma, osteolytic myeloma, and osteosclerotic myeloma.
- Osteosclerotic and osteolytic myelomas can usually be detected on a bone survey.
- “Skin changes” represent focal or generalized hyperpigmentation and/or skin thickening.
- Serum levels of VEGF normalize in response to therapy.
Amyloid disease
Can be sporadic or hereditary (see familial amyloid polyneuropathy below)
Tip:
MGUS is one of the few acquired neuropathies that is demyelinating. The most common causes of neuropathy tend to be predominantly axonal.
Toxins-related neuropathies
Alcohol-related peripheral neuropathy
- Classified as a toxic and not a nutritional neuropathy.
- While patients with chronic alcohol use are at risk of B12 deficiency and the associated peripheral neuropathy, studies have shown that chronic alcohol use can lead to an axonal neuropathy independent of B12 or B1 levels.
- EMG testing will show a sensorimotor axonal neuropathy with possible secondary demyelination.
Antibiotics
- The most common antibiotics to cause peripheral neuropathy are:
- Metronidazole, linezolid, and dapsone.
- It is also relatively common with chloroquine, fluoroquinolones, isoniazid, nitrofurantoin, and sulfasalazine
- The severity of metronidazole-related polyneuropathy is dependent on the cumulative dose and affects both large and small fibers.
- Recovery is often delayed for 6 to 12 months.
Chemotherapeutics
- Classically caused by platinum agents (cisplatin), vinca alkaloids (vincristine), and taxanes (paclitaxel, docetaxel).
- Risk increases in a dose-dependent manner.
Organic solvents and aerosols
- Acrylamide, carbon disulfide, hexane, toluene
- Seen with chronic exposure.
Heavy metals
- Lead, arsenic, mercury, thallium
- Chronic lead exposure presents with a motor-predominant neuropathy and can present with an ankle or wrist drop.
Autoimmune Neuropathies
Guillain-Barre syndrome (GBS)
- GBS represents an acute, monophasic immune-mediated polyneuropathy often preceded by infection (such as campylobacter jejuni, which causes a diarrheal illness) or other triggers (i.e. vaccine).
- While recognized as a single disease in the past, it is now known to represent a heterogeneous spectrum of both demyelinating and axonal disease patterns.
- Acute inflammatory demyelinating polyneuropathy (AIDP) is the most common form of GBS in the US (85-90%).
- Other variants include Miller Fisher syndrome, acute motor axonal neuropathy (AMAN), Acute motor and sensory neuropathy (AMSAN), and Bickerstaff brainstem encephalitis.
Acute inflammatory demyelinating polyneuropathy (AIDP)
- Commonly presents with acutely evolving symmetric weakness with hypoactive reflexes, starting distally and then extending proximally.
- Supportive features include mild sensory loss, a progression of symptoms up to 8 weeks, autonomic dysfunction, and possible cranial nerve involvement.
- Dysautonomia and respiratory muscle weakness with ventilatory failure are potentially serious complications
- Spirometry is the most effective measure of respiratory muscle function in these patients. Oxygen saturation is not a sensitive marker for impending respiratory dysfunction in neuromuscular disorders.
- Diagnostic testing:
- CSF studies will show an elevated CSF protein and normal WBC count (termed albuminocytologic dissociation). Although this is a peripheral nervous system disease, inflammation at the proximal nerve roots leads to an elevated protein in the CSF space.
- EMG/NCS: Early on, nerve conduction studies may all be normal except for prolonged F-waves. This occurs because the initial demyelination starts at the level of the nerve roots.
- Pathology: macrophage-mediated demyelination
- Anti-GM1 antibodies have been implicated in GBS, as well as multifocal motor neuropathy (MMN)
- Imaging: Contrast enhancement of the cauda equina and mild thickening of nerve roots.
- This pattern is non-specific and can be seen in conjunction with chronic inflammatory demyelinating polyneuropathy (CIDP), leptomeningeal carcinomatosis, granulomatous disease, or lymphomatosis.
- Pathology: macrophage-mediated demyelination
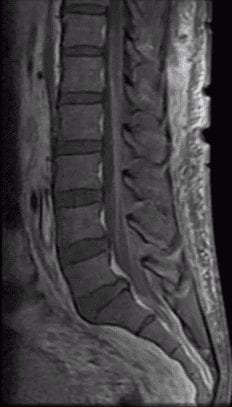

- Treatment: intravenous immunoglobulin (IVIG) or plasma exchange (PLEX). NOT steroids!
Miller Fisher syndrome
- Presents with the triad of ataxia, ophthalmoparesis, and areflexia.
- Associated with anti-GQ1b antibodies.
- Treatment is the same as AIDP.
Chronic inflammatory demyelinating polyneuropathy (CIDP)
- Patients present comparably to AIDP but have symptoms that progress longer than 8 weeks in duration.
- EMG/NCS will show markedly prolonged distal latency, temporal dispersion, conduction block, and decreased conduction velocities.
- Treatment: Steroids, plasmapheresis, and IVIG for acute management.
- Chemotherapeutics/immunosuppressive agents can be used for chronic use.
What you need to know
Steroids can be beneficial in CIDP but not AIDP!
Hereditary neuropathies
Charcot-Marie-Tooth disease (CMT)
- CMT is a class of hereditary peripheral neuropathies with various genetic causes
- They can be primarily demyelinating or axonal or both
- May also be called hereditary motor and sensory neuropathy (HMSN)
- There are over 30 causative genes related to CMT. Disease severity for each subtype can be quite variable. Below are the most high-yield variants:
CMT type 1A (CMT1A)
- Presents with weakness or sensory loss during the first two decades of life along with distal atrophy, hyporeflexia, palpable nerves, and high-arched feet.
- Patients will require ambulation aids such as ankle-foot orthoses but will not lose the ability to ambulate.
- Most common CMT phenotype.
- Due to the duplication of gene peripheral myelin protein (PMP22) on chromosome 17p (autosomal dominant inheritance).
- Pathology: Onion bulbs/hypertrophic neuropathy are the result of repeated episodes of demyelination and remyelination and are composed of concentric rings of Schwann cells.
- Onion bulbs can be seen in other chronic demyelination ⟷ remyelination processes too, like CIPD.
- EMG will show demyelinating sensory and motor peripheral neuropathy.
Reminder:
Deletion of the PMP22 gene can lead to hereditary neuropathies with pressure palsies (HNPP), while duplication can cause CMT type 1A.
CMT1B
- The second most common form of autosomal dominant CMT.
- The presentation can be similar to those CMT1 but more often patients will present with a severe early-onset form in infants.
- Affected children don’t walk until they are at least 15 months old.
- Severely affected patients will require significant assistance for ambulation (walkers, braces, motorized wheelchairs).
- Due to mutations of gene MPZ which encodes myelin protein zero, also known as P0 protein on chromosome 1.
CMT2
- Presents clinically similar to those with CMT1, however, EMG testing will show axonal neuropathy and not a demyelinating disease.
- Can be autosomal dominant or autosomal recessive.
*This is a high yield list of CMT phenotypes and far from an all-inclusive list.
Note:
Other than CMT2, which is an axonal neuropathy, all other “high yield” CMT variants are demyelinating. If EMGs are performed on demyelinating CMT variants it will show slow conduction velocities and prolonged latencies.
Hereditary neuropathy with pressure palsies (HNPP)
- Also known as tomaculous neuropathy.
- Tomacula: A pathological “sausagelike” swelling of myelin on peripheral nerves.
- This disorder is an autosomal dominant disease caused by a deletion or point mutation in the PMP22 gene.
- Symptomatic patients will present with isolated nerve palsies at common compression sites. Can also sometimes affect cranial nerves, namely CN VIII causing deafness.
- Hereditary neuropathy with pressure palsies may present in childhood but the onset and disease severity can be variable.
- Electrophysiologic studies reveal conduction blocks at sites of pressure and reduced motor and sensory conduction velocities.
- This can also be seen in asymptomatic gene carriers.
Hereditary transthyretin amyloidosis (hATTR)
- Formerly called familial amyloid polyneuropathy (FAP)
- Represents a group of autosomal dominant diseases that are due to mutations of transthyretin (TTR), apolipoprotein A-1, or gelsolin proteins.
- hATTR presents with length-dependent polyneuropathy and autonomic dysfunction.
- Treatment:
- Diflunisal (non-steroidal anti-inflammatory drug)
- Tafamidis (selective stabilizer of TTR)
- Patisiran (siRNA which reduces the production of TTR proteins)
- Liver transplant improves lifespan
- Treatment:
- Apolipoprotein A-1 can also have a similar neuropathy, but also presents with multiorgan (kidney, liver, GI) dysfunction due to amyloid deposition in these organs
- Gelsolin-related neuropathy is slow and fairly benign.
- hATTR presents with length-dependent polyneuropathy and autonomic dysfunction.
Fabry disease
- The most prevalent lysosomal storage disorder.
- Due to an X-linked defect in alpha-galactosidase which leads to the accumulation of globotriaosylceramide.
- Female carriers can have atypical, less severe phenotypes.
- Presents with a painful small fiber peripheral neuropathy with autonomic manifestations (anhidrosis).
- Other systemic signs/symptoms include: Angiokeratomas (reddish/blue rash) around the navel and the knees, renal disease, and left ventricular hypertrophy
- The risk of cerebrovascular disease is increased due to the accumulation of glycolipids in the endothelium of cerebral vessels.
Refsum disease
- Due to defective alpha-oxidation of phytanic acid.
- Patients present with peripheral neuropathy and palpable, enlarged nerves.
- Concurrent findings include ataxia, retinitis pigmentosa, and cardiac disease.
- Treatment: A diet low in branched fatty acids
- In severe cases, plasma exchange can lead to improvement of the neuropathy.
Ataxia-telangiectasia
- Due to mutations in the ATM gene.
- A mixed sensorimotor, length-dependent polyneuropathy develops before 10 years of age. However, it is usually not a predominant aspect of the disease since the other disease’s clinical features (ataxia) are more prominent and present earlier in life.
- Other systemic manifestations include ocular and dermal telangiectasias, diminished immunoglobulins, and malignancy.
Metachromatic leukodystrophy
- Associated with demyelinating sensorimotor polyneuropathy.
Friedrich’s ataxia (FA)
- Although not an early sign of the disease, patients develop peripheral sensory neuropathy leading to decreased vibration and joint position sense.
- Almost all patients will have sensory loss by the age of 25.
- Initial symptoms prior to sensory loss include progressive ataxia, and dysarthria/dysphagia.
- Other potential complications from FA include cardiomyopathy and diabetes.
Tangier’s disease
- Due to an ABCA1 gene mutation (autosomal recessive).
- This is an ATP cassette transporter protein.
- Patients present with sensory neuropathy, very low HDL, and orange tonsils.
References
- Biegstraaten, Marieke, et al. “Small Fiber Neuropathy in Fabry Disease.” Molecular Genetics and Metabolism, vol. 106, no. 2, 2012, pp. 135–141., doi:10.1016/j.ymgme.2012.03.010.
- Crawford, Thomas O. “Ataxia Telangiectasia.” Seminars in Pediatric Neurology, vol. 5, no. 4, 1998, pp. 287–294., doi:10.1016/s1071-9091(98)80007-7.
- Dick, D. J., et al. “Polyneuropathy in Hypothyroidism.” Postgraduate Medical Journal, vol. 59, no. 694, Jan. 1983, pp. 518–519., doi:10.1136/pgmj.59.694.518.
- Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31(2):491–510. doi:10.1016/j.ncl.2013.01.005
- Gemignani, Franco, et al. “Cryoglobulinemia Is a Frequent Cause of Peripheral Neuropathy in Undiagnosed Referral Patients.” Journal of the Peripheral Nervous System, vol. 7, no. 1, 2002, pp. 59–64., doi:10.1046/j.1529-8027.2002.02007.
- Harding, A. E. “Friedreich’s Ataxia: A Clinical And Genetic Study Of 90 Families With An Analysis Of Early Diagnostic Criteria And Intrafamilial Clustering Of Clinical Features.” Brain, vol. 104, no. 3, 1981, pp. 589–620., doi:10.1093/brain/104.3.589.
- Jameson, J. Larry, et al. Harrison’s Principles of Internal Medicine. McGraw Hill Education, 2018.
- Latronico, Nicola, and Charles F Bolton. “Critical Illness Polyneuropathy and Myopathy: a Major Cause of Muscle Weakness and Paralysis.” The Lancet Neurology, vol. 10, no. 10, 2011, pp. 931–941., doi:10.1016/s1474-4422(11)70178-8.
- Meché, F.g.a. Van Der, et al. “Diagnostic and Classification Criteria for the Guillain-Barré Syndrome.” European Neurology, vol. 45, no. 3, 2001, pp. 133–139., doi:10.1159/000052111.
- Mellion, Michelle, et al. “Alcohol-Related Peripheral Neuropathy: Nutritional, Toxic, or Both?” Muscle & Nerve, vol. 43, no. 3, 2011, pp. 309–316., doi:10.1002/mus.21946.
- Olney, Richard. “Neuropathies Associated with Connective Tissue Disease.” Seminars in Neurology, vol. 18, no. 01, 1998, pp. 63–72., doi:10.1055/s-2008-1040862.
- Planté-Bordeneuve, Violaine, and Gerard Said. “Familial Amyloid Polyneuropathy.” The Lancet Neurology, vol. 10, no. 12, 2011, pp. 1086–1097., doi:10.1016/s1474-4422(11)70246-0.
- Reilly, Mary M., et al. “Charcot-Marie-Tooth Disease.” Journal of the Peripheral Nervous System, vol. 16, no. 1, 2011, pp. 1–14., doi:10.1111/j.1529-8027.2011.00324.x.
- Robinson-Papp, Jessica, and Sonja G. Schütz. “HIV-Related Neuropathy: Current Perspectives.” HIV/AIDS – Research and Palliative Care, 2013, p. 243., doi:10.2147/hiv.s36674.
- Russell, James A. “General Approach to Peripheral Nerve Disorders.” CONTINUUM: Lifelong Learning in Neurology, vol. 23, no. 5, 2017, pp. 1241–1262., doi:10.1212/con.0000000000000519.
- Soubrier, Martin J., et al. “POEMS Syndrome: A Study of 25 Cases and a Review of the Literature.” The American Journal of Medicine, vol. 97, no. 6, 1994, pp. 543–553., doi:10.1016/0002-9343(94)90350-6.
- Thomson, Ruth M., and Gareth J. Parry. “Neuropathies Associated with Excessive Exposure to Lead.” Muscle & Nerve, vol. 33, no. 6, 2006, pp. 732–741., doi:10.1002/mus.20510.
- Toyooka, Keiko. “Fabry Disease.” Current Opinion in Neurology, vol. 24, no. 5, 2011, pp. 463–468., doi:10.1097/wco.0b013e32834a9433.
- Bhattacharyya, S., Darby, R., & Berkowitz, A. L. (2014). Antibiotic-induced neurotoxicity. Current infectious disease reports, 16(12), 448.
- Staff, Nathan P., et al. “Chemotherapy‐induced peripheral neuropathy: a current review.” Annals of neurology 81.6 (2017): 772-781.
- Surpur, Spurthi Sunil, and Raghav Govindarajan. “Role of “Sural Sparing” Pattern (Absent/Abnormal Median and Ulnar with Present Sural SNAP) Compared to Absent/Abnormal Median or Ulnar with Normal Sural SNAP in Acute Inflammatory Demyelinating Polyneuropathy.” Frontiers in neurology vol. 8 512. 29 Sep. 2017, doi:10.3389/fneur.2017.00512
Loading table of contents...
Loading table of contents...
Log in to View the Remaining 60-90% of Page Content!
Important: If you signed up after 1/1/2026, or if you opted to migrate your old account to the new & improved platform (same great content, better experience), please log in at nowyouknowmed.com
New here? Get started!
(Or, click here to learn about our institution/group pricing)1 Month Plan
Full Access Subscription
$142.49
$
94
99
1 Month -
Access to full question bank
-
Access to all flashcards
-
Access to all chapters & site content
3 Month Plan
Full Access Subscription
$224.98
$
144
97
3 Months -
Access to full question bank
-
Access to all flashcards
-
Access to all chapters & site content
1 Year Plan
Full Access Subscription
$538.47
$
338
98
1 Year -
Access to full question bank
-
Access to all flashcards
-
Access to all chapters & site content
Popular
