

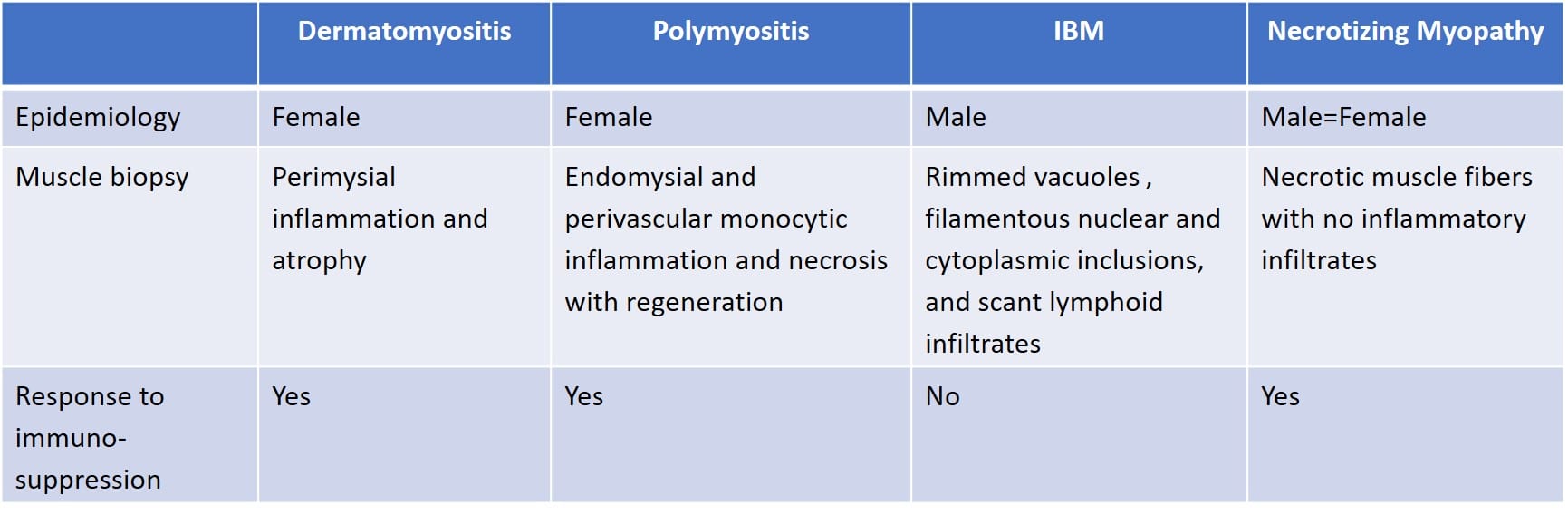
Idiopathic inflammatory myopathies
Polymyositis
- A female-predominant disease seen in one’s adult years that presents with subacute symmetric proximal weakness and pain.
- Can be associated with rheumatoid arthritis, HIV, or underlying malignancy.
- Muscle biopsy shows endomysial and perivascular monocytic inflammation and necrosis with regeneration.

Polymyositis
Note the inflammatory cells in the endomysium.

Polymyositis
Muscle biopsy with inflammatory cells in the endomysium (between and around individual myofibers) consistent with polymyositis.
Dermatomyositis
- A female-predominant disease which presents with subacute proximal weakness and pain.
- Dermatologic manifestations include a heliotrope rash on eyelids and an erythematous rash of the face or neck.
- Can be associated with connective tissue disease, malignancy, and interstitial lung disease.
- Muscle biopsy shows perifascicular inflammation and atrophy with sparing of the central fascicle.

Dermatomyositis
Note perimysial inflammation and atrophy.

Dermatomyositis
Trichrome stain with perifascicular atrophy.

Dermatomyositis
ATPase stain with perifascicular atrophy.
Inclusion body myositis (IBM)
A male-predominant slowly progressive idiopathic inflammatory condition of patients over the age of 50.
Clinical features include asymmetric weakness of the finger flexors and the quadriceps muscles.

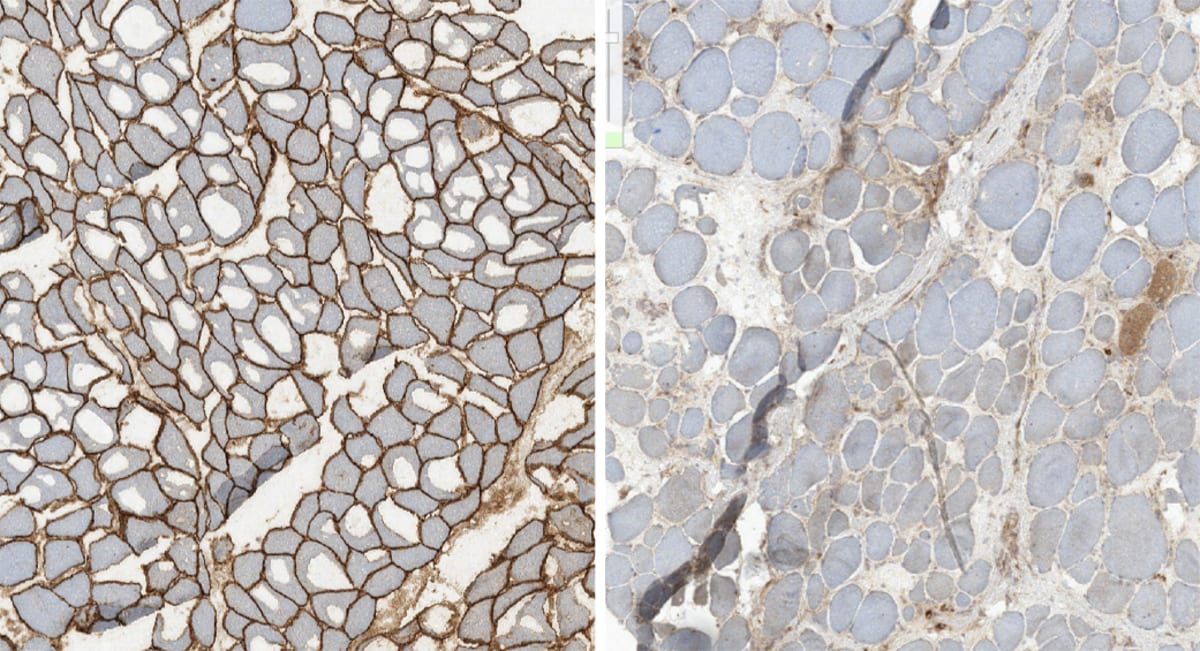
Inclusion Body Myositis
Note the rimmed vacuoles (arrow).

Inclusion Body Myositis
Note the rimmed vacuoles (arrow).

Inclusion Body Myositis
Note the lymphocyte invasion.
Treatment
- Polymyositis and dermatomyositis are responsive to immunosuppressive therapies (steroids, methotrexate, azathioprine, mycophenolate, etc.) while IBM is not.
Toxic myopathies
Steroid myopathy
- Occurs in the setting of chronic exposure.
- Patients present with progressive proximal muscle weakness with normal CK levels.
- EMG testing is typically normal.
- Discontinuation of steroid therapy leads to resolution of symptoms.
Statin-induced myopathy
- Less common than statin-induced myalgias, which can occur in as much as 20% of users.
- Pathogenesis is thought to be secondary to inhibition of mevalonic acid synthesis via inhibition of HMG-CoA reductase.
- Higher doses of statins and concurrent medications such as fibrates, niacin, calcium channel blockers, antiretrovirals, and cyclosporine can increase the risk of statin-induced myopathy.
Hydroxychloroquine-related myopathy
- Occurs in the setting of treatment for malaria or rheumatologic disease with hydroxychloroquine or chloroquine.
- Symptoms resolve with discontinuation of the medication.

Chloroquine Myopathy
Note the rounded fibers and vacuolar sarcoplasm.

Chloroquine Myopathy
Note granular amphophilic granules.

Chloroquine Myopathy
Electron microscopy showing curvilinear inclusions.

Chloroquine Myopathy
Acid phosphatase-positive stain.
Muscular dystrophies
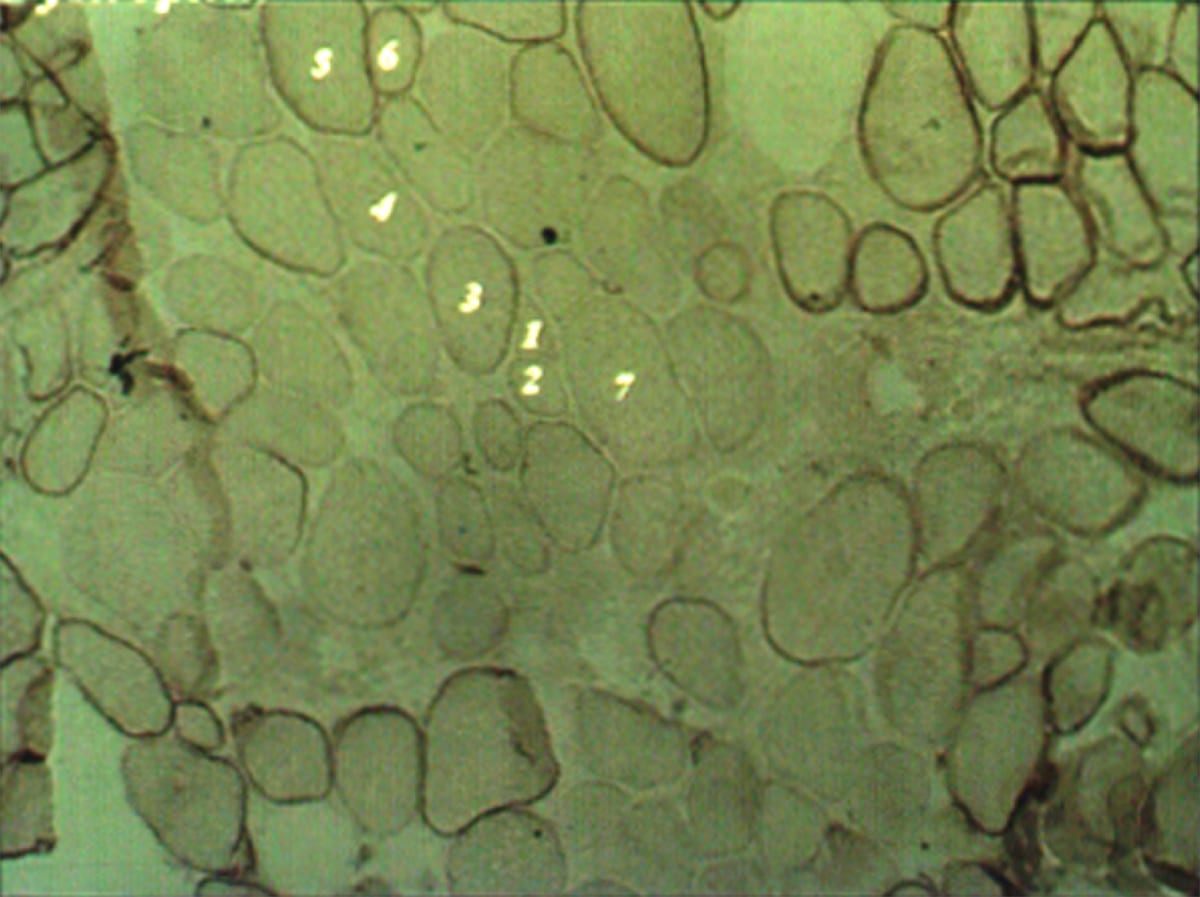
Duchenne muscular dystrophy (DMD)
- Due to an X-linked out-of-frame mutation of the dystrophin gene resulting in complete loss of dystrophin.
- Dystrophin anchors the cellular cytoskeleton to actin, contributing to the stability of the plasma membrane.
- Symptoms of weakness, cramping, and fatigue begin at 3-5 years of age with loss of ambulation by 13 years. Death secondary to respiratory or cardiac failure typically occurs in the second to third decade of life.
- Exam on initial presentation will show pseudohypertrophy in calf muscles, a waddling gait, toe walking, and Gower’s sign.
- Pathology: Degenerating fibers of various sizes undergoing phagocytosis, excessive fibrosis, and connective tissue.
- Immunohistochemical staining for dystrophin will show a complete lack of staining.
- Treatment:
- Corticosteroids slow the progression of the disease.
- Eteplirsen, which works by exon skipping of dystrophin mRNA, was recently FDA-approved for a subset of patients with DMD who have out-of-frame mutations on exon 51 of the dystrophin gene.
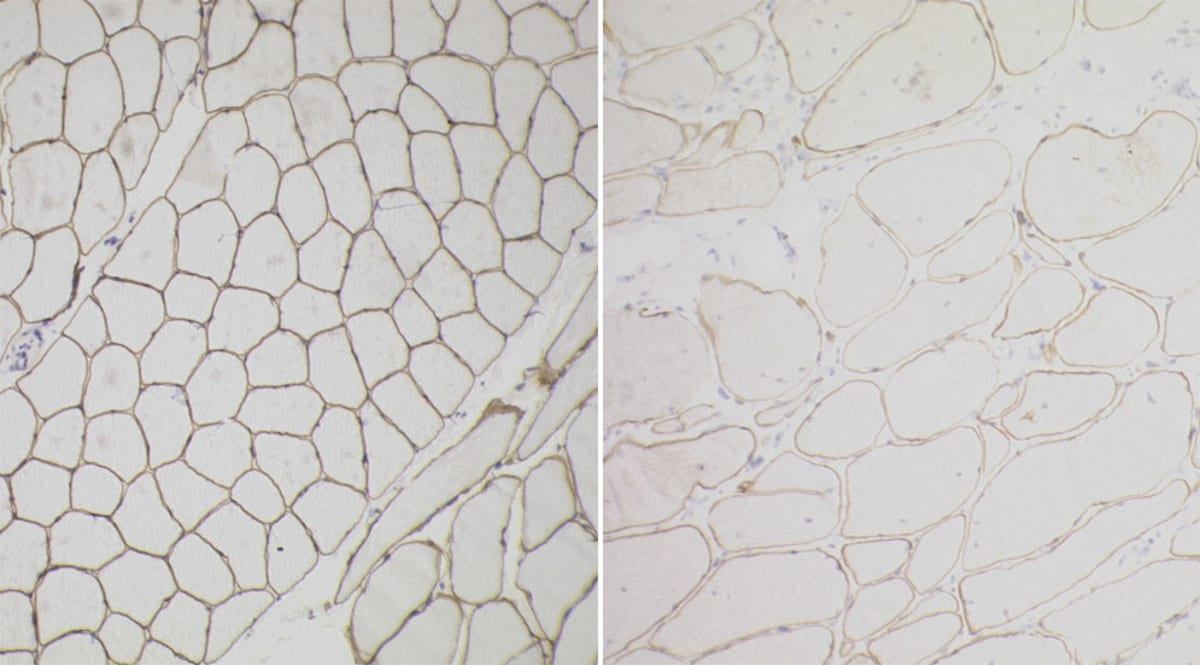
Becker muscular dystrophy
- Patients present similarly to DMD but will do so later in life due to the partial function of dystrophin via an in-frame mutation of the dystrophin gene.
- Immunohistochemical staining for dystrophin will show partial staining.
- Treatment: corticosteroids
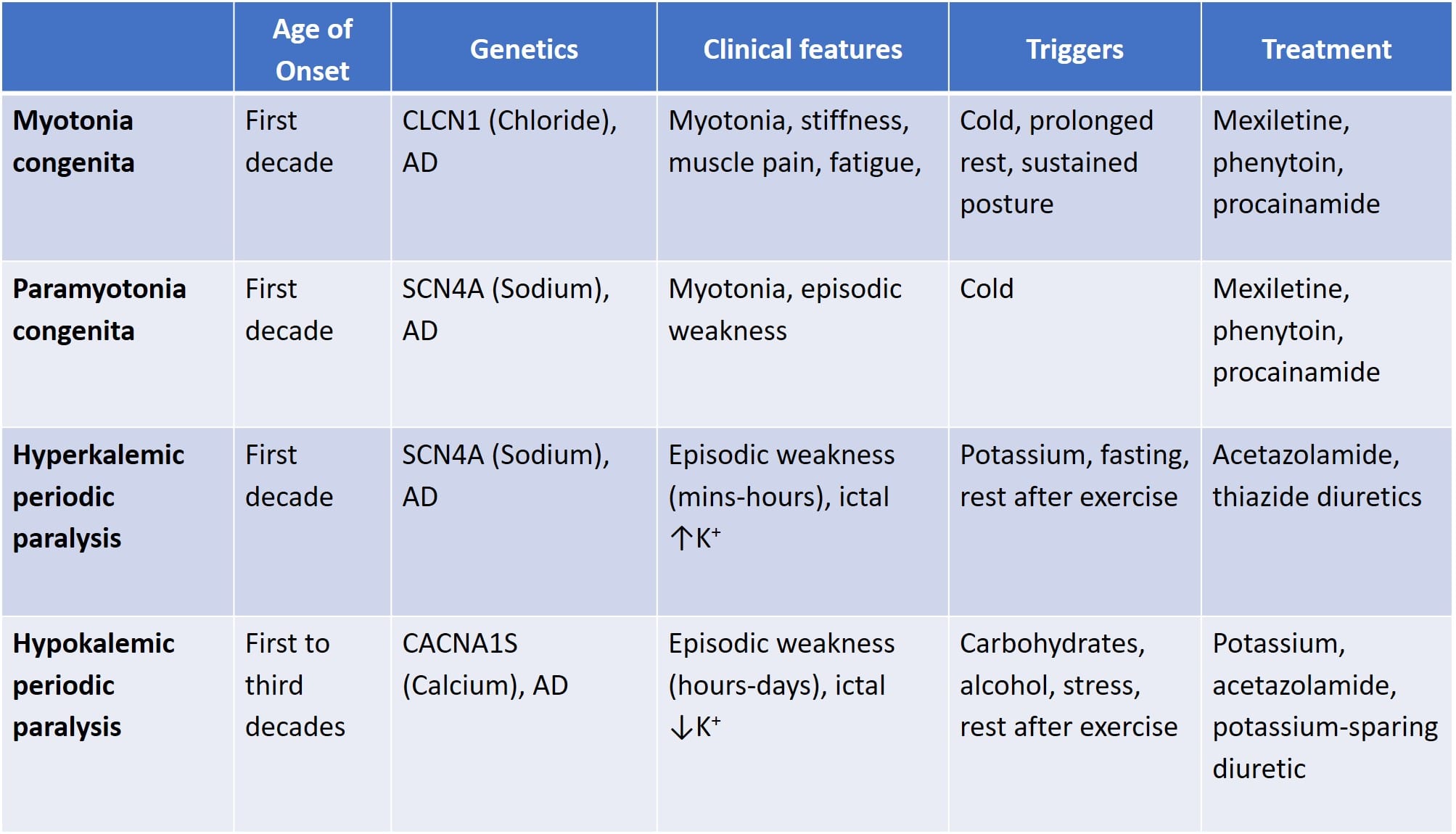
Myotonic dystrophy (MD)
- The most common muscular dystrophy in adults.
- Type 1 MD is an autosomal dominant disorder due to a CTG repeat of the myotonic dystrophy protein kinase (DMPK) gene that anticipates with successive generations.
- Type 2 MD is also autosomal dominant, but due to a CCTG repeat of the CNBP gene.
- Patients present with myotonia as well as weakness of the face, neck, and intrinsic hand muscles.
- Myotonia can be treated with sodium channel blockers (mexiletine, phenytoin, and carbamazepine) and tricyclic antidepressants (amitriptyline, clomipramine, and imipramine).
- Other symptoms include excessive daytime sleepiness, conduction defects, cataracts, frontal balding, ptosis, and endocrine dysfunction.
- Patients with MD need to have interval EKGs for cardiac arrhythmias.
- EMG will show myotonic discharges (spontaneous potentials with waxing and waning amplitude and frequency) on needle EMG.
Fukuyama muscular dystrophy
- Presents in the neonatal period with hypotonia, contractures, and gross motor delays
- Cerebral dysgenesis and intellectual disability are often seen as well.
- Associated with mutations to fukutin (putative glycosyltransferase) gene.
EMG findings in muscular dystrophies:
- Fibrillation potentials, positive sharp waves, and short, small, polyphasic MUAPs with early recruitment.
Congenital myopathies
Myotubular/centronuclear myopathy
- A disease that presents with hypotonia, weakness involving the cranial nerve innervated muscles, and cognitive changes.
- Pathology:
- Muscle biopsy will show centrally-located nuclei with a perinuclear sarcoplasmic reticulum extending radially in a “halo” formation.
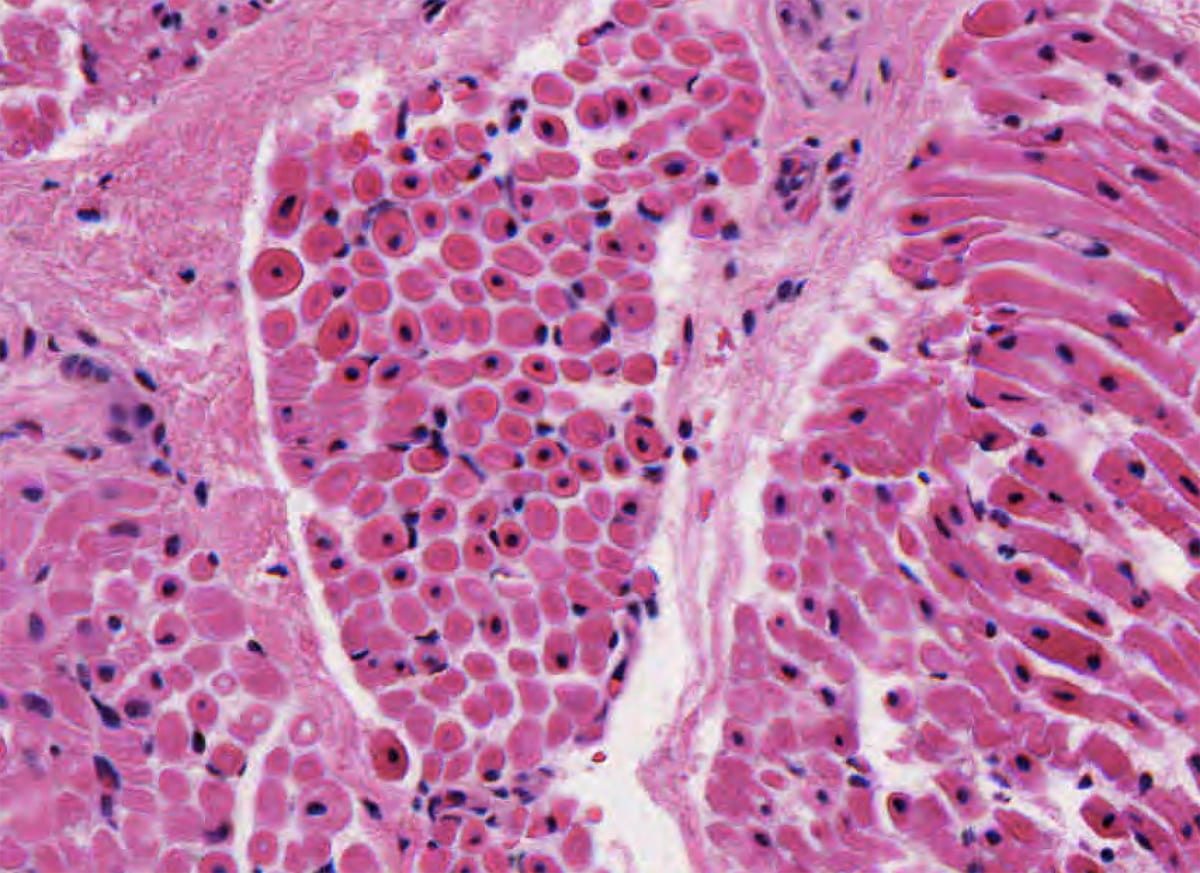
Nemaline myopathy
- Presents with infantile hypotonia.
- Pathology:
- Muscle biopsy will show a finding of rod- or thread-like eosinophilic structures on Gomori trichrome stain.
- Electron microscopy shows rod-like or oval, electron-dense bodies radiating from the sarcomeric Z-line.
Central core disease
- An autosomal dominant disease that presents with neonatal hypotonia and weakness secondary to mutations to the ryanodine calcium channel (RYR1).
- Pathology:
- Muscle biopsy will show lucent central cores on NADH stain with variably sized fibers with internalized nuclei.
- Patients are at a higher risk to develop malignant hyperthermia.
Mitochondrial myopathies
- An umbrella term that includes several syndromes such as myoclonic epilepsy with ragged red fibers (MERRF), mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) .
- Pathology:
- Ragged red fibers are typically seen on muscle biopsy. Other findings include increased subsarcolemmal staining on succinate dehydrogenase (SDH) stains (ragged blue fibers).

Mitochondrial Myopathy
Trichrome stain showing ragged red fibers.

Mitochondrial Myopathy
Trichrome and gomori stain showing ragged red fibers.

Mitochondrial Myopathy
SDH stain showing ragged blue fibers.
MELAS
- “Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes”
- The name alone gives you most of the facts needed for neurology examinations.
- Associated with mutations related to mitochondrial MT-TL1.
MERRF
- “Myoclonic epilepsy with ragged red fibers”
- The name alone gives you most of the facts needed for neurology examinations.
Log in to View the Remaining 60-90% of Page Content!
Important: If you signed up after 1/1/2026, or if you opted to migrate your old account to the new & improved platform (same great content, better experience), please log in at nowyouknowmed.com
