

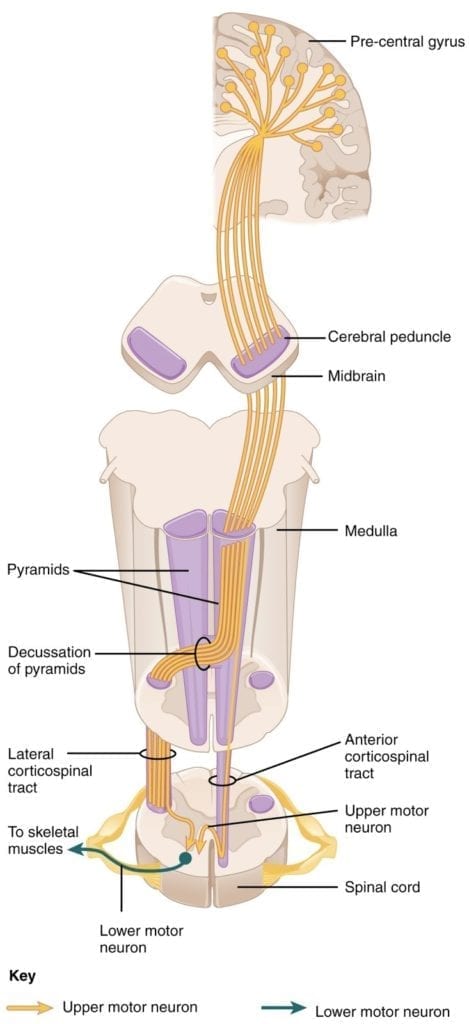
- The major descending tract that controls skeletal muscle movements is the corticospinal tract. It is composed of two neurons: the upper motor neuron and the lower motor neuron. Upper motor neurons have their cell bodies in the primary motor cortex of the frontal lobe and their axons travel between the basal nuclei and caudate nucleus as the internal capsule. These axons then traverse through the midbrain as the cerebral peduncle and then decussate upon entering the medulla.
- Upper motor neuron axons continue in the spinal cord as the corticospinal tract, then synapse on lower motor neurons in the ventral horn of the spinal cord. Lower motor neuron axons then project to skeletal muscle in the periphery (Figure 1).

Figure 1: Corticospinal tract. Courtesy of OpenStax College, Anatomy & Physiology.
- ALS is a progressive degenerative disorder that causes denervation of the striated muscle.
- Patients will present with both upper (hyperreflexia, spasticity) and lower motor neuron signs (fasciculations, muscle atrophy). Symptoms are often asymmetric at the onset.
- Bulbar onset ALS can also occur 25% of the time.
- Commonly presents in the 5th or 6th decades of life.
- Progressive symptoms usually lead to respiratory failure and death within 3-5 years.
- Older age at symptom onset, early respiratory dysfunction, and bulbar onset of symptoms have a worse outcome.
- ALS is often sporadic, but 5-10% of cases are familial.
- C9ORF72 (GGGGCC hexanucleotide repeat in a noncoding region)
- Accounts for 40% of familial ALS (most common cause).
- Families will also present with concurrent frontotemporal dementia.
- SOD1 mutation (chromosome 21): encodes for copper/zinc ion-binding superoxide dismutase.
- Accounts for 20% of familial ALS.
- Others include TDP-43 and FUS
- C9ORF72 (GGGGCC hexanucleotide repeat in a noncoding region)
- While ALS is a clinical diagnosis, definitive diagnosis is made after EMG/NCS shows denervation .
- Sensory nerve conduction studies are typically normal.
- Pathology:
- On myelin stain cross-sections of the spinal cord, pallor and Wallerian degeneration of the cortical spinal tracts can be appreciated. (Image 1)
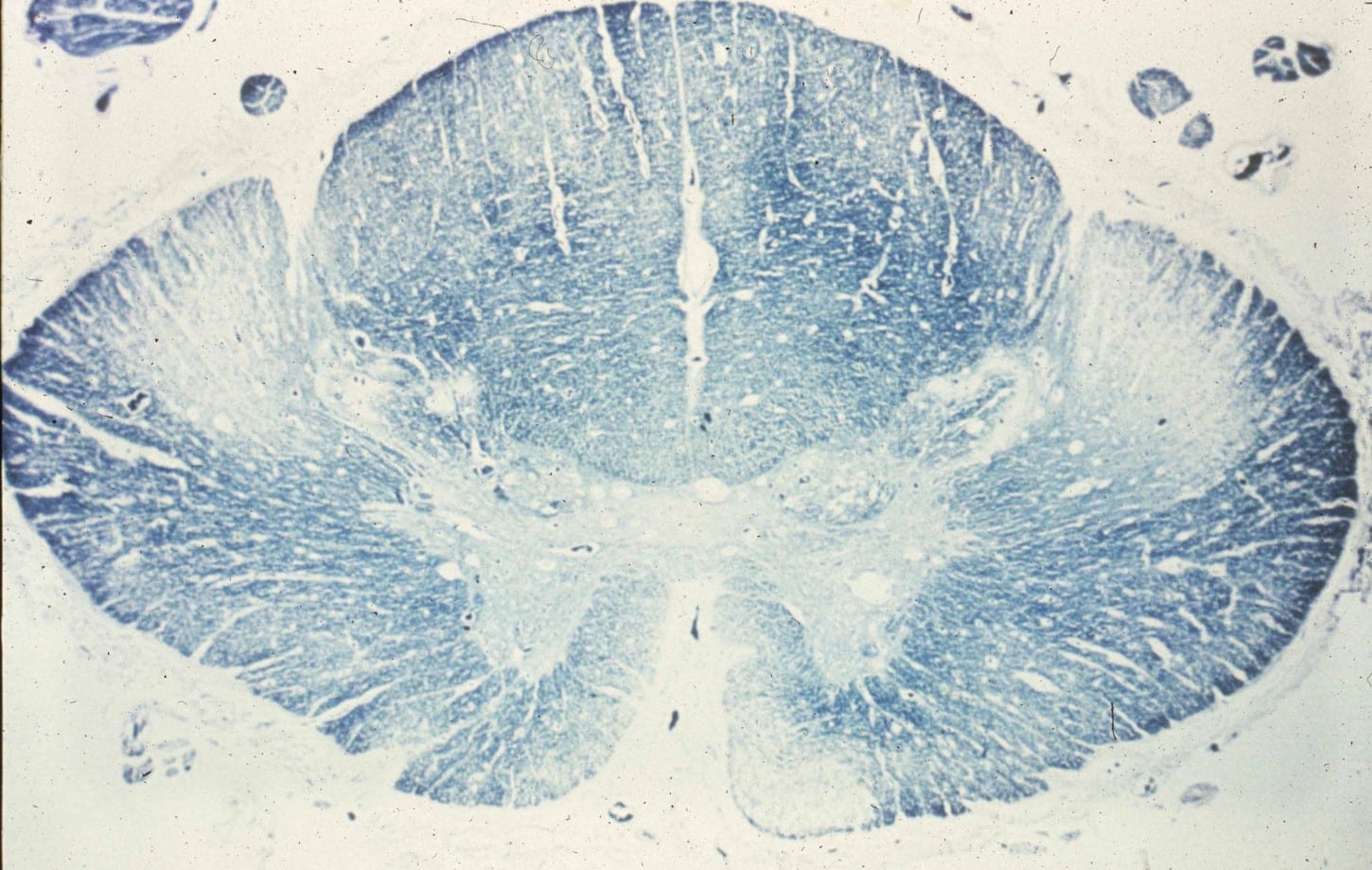
- Microscopic findings:
- Inclusion bodies can be found in anterior horn cells.
- TDP-43 inclusions
- Seen in most cases of ALS.
- TDP-43 protein is an RNA binding protein involved with transcription regulation
- When abnormally phosphorylated and ubiquitinated it leads to various inclusion types (neuronal intranuclear, neuronal cytoplasmic, glial inclusions, etc).
- TDP-43 inclusions are also seen in half of patients with FTD.
- Treatment:
- Riluzole: A glutamate release inhibitor that prolongs survival 2-3 months on average. Common side effects include fatigue and GI issues.
- Edaravone: FDA approved in 2017, edaravone is a free-radical scavenger shown to inhibit motor neuron death by reducing oxidative stress.
- ALS variants
- Primary lateral sclerosis: Presents with UMN symptoms only.
- Progressive muscular atrophy: Presents with LMN symptoms only.